

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Sarcopenia and osteoporosis have recently become increasingly significant as the population of older people increases and are clinically very important as common pathological states. In skeletal tissues, muscle and bone interact mechanically and functionally. Numerous lines of evidence suggest the remote interactions between muscle and bone as well as their local interactions. Genetic, endocrine, mechanical and age-related factors influence both muscle and bone simultaneously. However, the physiological and pathological mechanisms related to both muscle and bone still remain unclear, although interest in muscle/bone relationships as well as muscle biology has been increasing recently. In this review, I describe several aspects of the interactions between muscle and bone.
1. Sarcopenia and osteoporosis
Aging and various pathological states influence muscle and bone simultaneously. Sarcopenia is a condition involving decreases in muscle mass and function, which is related to frailty. Sarcopenia may lead to physical function abnormality, decreased quality of life and increased mortality of patients. It is common in older people, with a reported prevalence in 60- to 70-year-olds of 5-13% and a prevalence in those >80 years old of 11-50%. Sarcopenia is also significantly associated with osteopenia and osteoporosis in Japanese women.[1]
The mechanism of concomitant bone and muscle loss with aging is not clear at present. Muscle mass decline with aging appears to occur before bone mass decline with aging. Greater adiposity is increasingly observed in both bone marrow and muscle, and fat infiltrates are also observed in nerves and capillaries. Neurological mechanisms may also be related to common deficits in physical performance attributed to sarcopenia and osteoporosis.[2]
2. Bone and body composition
Higher body mass index (BMI) is related to higher bone mineral density (BMD) and reduced fracture risk. The mechanism is presumably due to an increased strain on bone imposed by higher body mass, estrogen production from the greater amount of adipose tissue and the cushioning defense of the hip by gluteofemoral adipose tissue, which reduces impact forces upon falling. Hip fracture risk was found to be increased with decreasing BMI independently of physical activity in a large prospective study on postmenopausal women. In addition, in a cross-sectional study in the United Kingdom, obese patients were more likely to suffer fractures at the ankle and upper arm, suggesting that higher BMD in obesity is not protective against fractures, which might be related to body habits, mechanisms of injury and the effects of adiposity on bone.[3]
3. Relationships between muscle and bone in clinical studies
Lean body mass (LBM) is related to BMD in elderly men, and it explained 20% of the variability in BMD at the femoral neck.[4] Repetitive loading exerted exercise-induced benefits on bone mass and muscle area in a yearlong study of 10- to 17-year-old tennis players.[5] The change of muscle area explained 32% of the variability in the exercise-induced benefits in bone mass, which seemed to be higher than that in postmenarcheal girls.
Controversy exists as to whether higher fat mass positively or negatively affects fracture risk. Recent evidence suggests that higher fat mass might be related to increased fracture risk, although it positively affects BMD. Although body weight has increased dramatically in older people in Western countries and Asia, many, if not most, osteoporotic fractures occur in overweight or obese people, and obese men may be particularly susceptible.[6,7] This may be due to lower physical activity induced by obesity, leading to disability or institutionalization. Alternatively, adipocytokines produced from adipose tissues might negatively affect bone to increase fracture risk.
Muscle parameters are related most strongly to cortical area and total shaft area, but explained <10% of variability in those bone parameters in mid-thigh computed tomography analysis, and small muscle area as well as low cortical thickness was significantly associated with fractures in both sexes.[8] However, this study suggested that bone and muscle loss proceed at different rates with aging and sex-related patterns.
Numerous studies indicate that higher LBM is related to increased BMD and reduced fracture risk, especially in postmenopausal women.[9] Age-related sarcopenia is affected by two components for diagnosis: low muscle mass and function.[10] Decreased muscle mass does not often parallel functional disability. Muscle mass and muscle strength were also independently associated with postmenopausal osteoporosis, and they should be considered separately in clinical practice.[11] In addition, middle-aged and elderly community-dwelling European men with reduced muscle mass had significantly lower BMD and higher prevalence of osteoporosis.[4]
Weight loss therapy to improve health in obese older adults causes further bone loss. The addition of exercise training to weight loss therapy among obese older adults prevented weight loss-induced increase in bone turnover and attenuated weight loss-induced reduction in hip BMD, and the change in LBM was one of the independent predictors of change in hip BMD in that study.[12] The increase in sclerostin levels with weight loss was also found to be prevented by exercise in obese older adults, and an inverse relationship was found between the changes in sclerostin and LBM.[13] Since sclerostin suppresses the canonical Wnt-β-catenin signal, which inhibits muscle differentiation, sclerostin may be related to exercise-induced changes of muscle and bone through its production from osteocytes, which induces sensitization to the mechanical signal.
In another study, long-term body composition changes were followed for 6 years in French women.[14] LBM and fat mass did not change in premenopausal and perimenopausal women. However, LBM and bone mass decreased, but fat mass increased in postmenopausal women. Age was the most important determinant of body composition changes, although menopausal status was a significant determinant only for the changes in bone mass. The comparisons of cross-sectional versus longitudinal associations of LBM and fat mass with BMD in children showed that cross-sectional associations for LBM and fat mass with bone may not reflect longitudinal associations.[15]
An increase in muscle mass produces stretching of collagen fibers and periosteum at the interface, resulting in the stimulation of local bone growth. Alternatively, higher blood flow to bone might lead to an increase in bone strength, since blood flows to limbs at a level proportional to muscle mass.
As for the relationships between muscle and treatment, high-frequency, low-intensity vibrations increased bone mass and muscle strength in upper limbs in a prospective clinical trial on 65 disabled children.[16] A recent study also indicated that low appendicular muscle mass of the upper limbs and low grip strength are related to poor cortical and trabecular microarchitecture, partly independently of each other, in older men.[17] The associations were significant after adjustment for confounders including body size.
4. Bone and muscle interactions during development
A close relationship between bone and muscle is observed during development and growth. Several studies suggest that the Indian Hedgehog pathway and fibroblast growth factor (FGF)-2 may play important roles in the interactions between muscle and bone during development.[2] The peak velocity for LBM precedes that of BMD, indicating that an increase in muscle mass during growth stimulates the increase in bone mass.[18] Circulating insulin-like growth factor (IGF)-I promotes bone mass accrual during puberty, although muscle secretes IGF-I as one of the potential sources.[2]
5. Genetic factors
Since both bone and muscle cells are derived from mesenchymal stem cells, similar genetic factors are considered to influence bone and muscle. Risk factors affecting osteoporosis and sarcopenia are heritable at approximately 60-70% heritability.[19] Osteoporosis and sarcopenia may be affected by genetic polymorphisms of several genes, such as androgen receptor, estrogen receptor, catechol-O-methyltransferase, IGF-I, vitamin D receptor and low-density-lipoprotein receptor-related protein 5.[19] In a young adult twin study, the relationship between LBM and BMD was shown to be influenced by genetic factors, compared with the relationship between fat mass and BMD.[20] Several genes, such as growth and differentiation factor-8 (GDF-8), myocyte enhancer factor-2C (MEF-2C) and proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), have also been detected in genome-wide association studies as being linked to both sarcopenia and osteoporosis.[21]
6. Endocrine factors
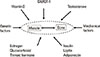
There are numerous physiological and pathological endocrine factors that influence both muscle and bone (Fig. 1). Vitamin D, the growth hormone (GH)/IGF-I axis and testosterone are the most important hormones that affect muscle and bone simultaneously. Moreover, estrogen, glucocorticoid, thyroid hormone, insulin, leptin and adiponectin also regulate muscle/bone relationships, as described in the following paragraphs.
Other major factors may affect both muscle and bone negatively, which include nutritional state, physical activity, atherosclerosis, hormones and postinflammatory cytokines.[10]
7. Vitamin D
Vitamin D exerts various effects on bone and muscle cells. Vitamin D insufficiency is very common in elderly people. The measurement of serum 25-hydroxyvitamin D (25[OH]D) level is recommended as an initial diagnostic test in patients at risk of vitamin D deficiency.[22] Vitamin D deficiency is defined as 25(OH)D below 20 ng/mL (50 nmol/L) in the guidelines. A recent systematic review showed that the average increase in serum 25(OH)D concentration was 0.78 ng/mL (1.95 nmol/L) per microgram of vitamin D3 supplement per day in the absence of concomitant use of calcium supplements.[23] Post-hip fracture use of prescribed calcium plus vitamin D or vitamin D supplements and antiosteoporotic drugs seemed to lead to lower mortality in both sexes, although vitamin D alone did not seem to be effective in the elderly.[24,25]
Human skeletal muscle has a receptor for 1,25(OH)D, and vitamin D receptor genotype variations affect BMD and muscle strength. The changes of muscle fibers as well as muscle differentiation-related genes, such as Myf-5, myogenin and E2A, occur independently of calcium metabolism in vitamin D receptor-deleted mice. Severe osteopenia and sarcopenia are observed in patients with vitamin D-deficient osteomalacia. A higher prevalence of atrophy among type II fibers in osteoporotic patients with low levels of 25(OH)D has been reported. Vitamin D deficiency causes increased risk of falls through the effects of vitamin D deficiency on bone as well as through its effects on muscle, and vitamin D supplementation reduces the risk of falls in vitamin D-deficient patients. An increased risk of falls is directly linked to fractures. However, intermittent large doses of vitamin D (oral cholecalciferol at 150,000 IU every 3 months) were not effective for falls, mobility and muscle strength in older postmenopausal women.[26] Marantes et al.[27] also reported that low 25(OH)D or high parathyroid hormone (PTH) levels did not contribute significantly to sarcopenia or muscle weakness in community adults. This study suggests the age-dependent differences between vitamin D state and sarcopenia.
The interrelationships between muscle and bone related to vitamin D action and the molecular mechanisms by which vitamin D affects both bone and muscle are unclear at present. A recent study revealed that 1,25(OH)D3 induces myogenic differentiation by inhibiting cell proliferation, decreasing IGF-I expression, promoting myogenic differentiation through increasing IGF-II and follistatin expression, and by decreasing myostatin.[28]
8. GH/IGF-I axis
GH as well as IGF-I induces muscle hypertrophy as well as bone development and the preservation of bone mass. GH deficiency causes reduction in muscle and bone mass and an increase in fat mass. Several studies indicate that serum IGF-I levels are positively related to LBM and a reduction of fracture risk. Thus, the GH-IGF-I axis is one of the crucial pathways for the maintenance of bone mass and strength. IGF-1 enhances the proliferation of muscle progenitor cells and their integration with existing fibers during muscle repair. The level of mechano growth factor (MGF), which is derived from the IGF-I gene by alternative splicing, declines with aging, and MGF administration activates muscle stem cells that are important for muscle repair and hypertrophy.[29] Osteoporosis was found to be associated with a preferential type II muscle fiber atrophy, which correlated with BMD and reduced the level of Akt, a component of the IGF-I/PI3kinase/Akt pathway, in a muscle biopsy study of older people, although muscle atrophy was less related to disease duration and severity in osteoarthritis.[30]
In female-to-male transsexuals after long-term cross-sex hormonal therapy after ovariectomy, the transsexual men on long-term testosterone therapy demonstrated higher muscle mass and greater grip strength as well as lower fat mass and increased trabecular BMD, although there were a larger radial cortical bone size and lower cortical volumetric BMD at the radius and tibia in these men.[31] These data suggest that testosterone and estrogen differently affect muscle and bone, and that testosterone may mainly affect bone size, but not BMD, partly through muscle factors in cortical bone. Although raloxifene, a selective estrogen receptor modulator, is effective for the treatment of osteoporosis, GH cotreatment with 17β-estradiol increased LBM and BMD at the lumbar spine and femoral neck to a greater extent than raloxifene in hypopituitary women.[32] These findings suggest that raloxifene significantly attenuates the beneficial effects of GH on body composition.
IGF-binding proteins (IGFBPs) play some role in the GH-IGF-I axis dependently and independently of IGF-I. Appendicular skeletal muscle mass (ASM) was found to be associated with cortical thickness and trabecular BMD in a cohort study.[33] In that study, serum IGFBP-2 levels were the most robust negative predictors of ASM in both sexes and might provide new insights into potential biomarkers that reflect the health of the musculoskeletal system.
9. Sex hormones
Estrogen and testosterone regulate bone and muscle simultaneously. Androgens play a significant role in the development and maintenance of muscle and skeletal integrity in both men and women. Testosterone levels are correlated with BMD and muscle strength. Androgen deficiency is characterized by loss of bone and lean tissue.[21] Although skeletal muscle is one of the most powerful determinants of bone strength, sex differences in the bone-muscle relationship may be important for explanations of sex differences in bone growth, age-related bone loss and fracture risk.[34]
In young adulthood, there are apparent sex differences in the correlation of muscle area to bone area. More of the variation in bone dimensions is explained by muscle area in men. Women have higher values of bone in relation to muscle, but a lower percentage of the variation in cortical area in women is explained by muscle mass.[34] Higher endogenous free testosterone levels are associated with higher BMD, greater LBM and greater fat mass in women aged 65 and older.[35] These findings suggest the possibility that testosterone or selective androgen receptor modulator might be expected as a drug for the treatment of both sarcopenia and osteoporosis in women as well as men.
10. Glucocorticoid excess and diabetes
Glucocorticoid is used for the treatment of patients with rheumatic, hematologic, neurologic and chronic pulmonary diseases. Simultaneous negative influences of glucocorticoid excess for Cushing's syndrome or its exogenous administration on both muscle and bone are well known. Glucocorticoid excess induces an increase in fracture risk, especially at trabecular bone and in elderly patients, through decreased bone quality as well as decreased BMD. However, how glucocorticoid excess affects the interactions between muscle and bone is still unknown. Our previous study revealed that femoral neck BMD was negatively related to percent LBM in postmenopausal women with glucocorticoid treatment, although the influence of body composition on vertebral fracture risk seemed to differ depending on age.[36] Moreover, glucocorticoid use was independently related to 25(OH)D deficiency in a large, nationally representative sample of children and adults,[37] although vitamin D deficiency affects both muscle and bone.
Diabetes is also an important causal disease for secondary osteoporosis. Although osteopenia and severe increase in bone fragility are known in type 1 diabetes, numerous recent studies indicate that fracture risk is increased in type II diabetes, presumably via a decrease in bone quality, sarcopenia and an increased risk of falls. Proximal dominant myopathy is observed in some diabetic patients, and a preferential and diffuse involvement in type II fibers has been described. Skeletal muscle in type 2 diabetes is characterized by insulin resistance, impaired glycogen synthesis, impairments in mitochondria and lipid accumulation. Bone quality in type 2 diabetes is decreased, potentially due to the effects of advanced glycation end-products on collagen, impaired osteoblast activity and lipid accumulation. Muscle density was also found to be positively related to physical activity and negatively associated with markers of fat distribution and risk for type 2 diabetes, when fat and muscle indices were assessed by peripheral quantitative computed tomography at forearm and foreleg.[38]
Body weight control and exercise therapy as well as drug therapy for diabetes modulate the interactions between muscle and bone. Although body weight control may reduce both muscle and bone mass in diabetic patients, one year of an intensive lifestyle intervention in adults with type 2 diabetes along with weight loss was related to a modest increase in hip bone loss despite improved fitness and glycemic control.[39] Loss of muscle mass by diet therapy should be paid attention for the treatment of diabetic patients. Resistance exercise involves the movement of high loads using resistance from either machines or weights for a smaller number of repetitions, although aerobic exercise is recommended as the usual exercise therapy for diabetes. Several studies suggest that resistance training (strength training) may impose potent and unique benefits in type 2 diabetes by treating the dysfunction of both muscle and bone induced by diabetic metabolic abnormalities.[40]
11. Mechanical factors
Mechanical stress changes, such as immobilization and lack of gravity, greatly influence both muscle and bone. Astronauts lose both muscle and bone mass. Muscle loss is recovered about six months faster than bone loss in astronauts.[41] Several lines of evidence have shown that low-magnitude mechanical signals are anabolic to bone and muscle.[42,43] Clinical studies also suggested that low-intensity vibration signals stimulate bone and muscle formation as well as increase muscle force activity. They stimulate mesenchymal stem cell proliferation and bias their differentiation toward osteoblastogenesis and away from adipogenesis,[44] suggesting that fate selection in hematopoietic progenitors can be determined by mechanical signals.
12. Muscle and bone relationships
Several studies have indicated that higher muscle mass is closely related to increased BMD and reduced fracture risk in postmenopausal women. Calcium ions are also critical for muscle contraction, and hypocalcemia induces muscle tetany. In addition, muscle and bone are simultaneously influenced by pathological states, such as glucocorticoid excess and vitamin D deficiency. These findings raise the possibility that there might be interactions between muscle and bone metabolism.[9]
Fractures that are covered with relatively intact muscle were found to improve more rapidly than fractures associated with more severe damage. Muscle flaps applied to autogenous bone grafts also improved healing. Proinflammatory cytokines, in particular tumor necrosis factor (TNF)-α, at the site of fracture induced the differentiation of stromal cells present in muscle into osteoprogenitor cells and promoted bone fracture healing.[45] A recent study also demonstrated that muscle-derived stem cells take on a primary role in the reparative response in the setting of severe injury to the periosteum.[44] These findings suggest that muscle tissues play important physiological and pathological roles through certain interactions between muscle tissues and bone metabolism.
13. Disease linking muscle to bone
Fibrodysplasia ossificans progressiva (FOP) is an important clinical clue as a disease linking muscle to bone.[9] It is a rare autosomal dominant disorder with skeletal malformations and progressive extraskeletal ossification. Heterotopic ossification of the muscles, tendons, ligaments and fascia begins in childhood and can be induced by trauma or for no clear reason, leading to extra-articular ankylosis of all major joints in the axial and appendicular skeleton, which renders movement impossible.
A heterozygous constitutively activating mutation (R206H) in bone morphogenetic protein (BMP) type I receptor, the activin receptor type I (ACVR1/activin-like kinase 2 [ALK2]), is found in patients with the classic form of FOP. Constitutive activation of the BMP signaling molecule Smad1 or Smad5 induces ectopic bone formation in FOP. These findings indicate that constitutive activation of BMP signaling by the ALK2 mutation is responsible for the molecular pathogenesis of FOP. We reported that serum from a patient with FOP includes some soluble factors that might enhance osteoblast differentiation and BMP-2 expression in mouse osteoblastic cells.[46] Middle-age onset of heterotopic ossification was reported in a case of FOP with the mild alteration of ALK2 from a unique missense mutation (G325A).[47] BMP-9 is involved in the pathophysiology of heterotopic ossification, with its activity depending on the skeletal muscle microenvironment, such as damage.[48] Overactive BMP signaling is involved in the pathogenesis of heterotopic ossification and Duchenne muscular dystrophy due to a mutation of the dystrophin protein that connects the cytoskeleton of muscle fibers to the underlying basal lamina,[49] although ALK3, a BMP receptor, is involved in the muscle regeneration process. BMP signaling in the satellite cells may exacerbate the disease in Duchenne muscular dystrophy.
14. Local factors affecting muscle ossification
The details of the heterotopic ossification of muscle in FOP remain to be fully elucidated. Since ossification does not occur in muscle tissues in the physiological state, there might be some local regulators that enhance or suppress ossification specifically in muscle tissues. We hypothesized that further study of the ALK2 (R206H) mutant receptor should lead to additional insights into the ossification of muscle. We therefore performed a comparative DNA microarray analysis between empty vector- and ALK2 (R206H)-transfected mouse myoblastic C2C12 cells.[50] Several bone-related factors, such as Tmem119, osteoactivin and Frizzled-3, were induced by ALK2 (R206H) overexpression (Fig. 2). Among them, Tmem119 is a PTH-responsive Smad3-related factor, interacting with Smad1/5 and Runx2 in osteoblastic differentiation.[51]
We demonstrated that Tmem119 promotes the differentiation of myoblasts into osteoblasts, suggesting that it may play a critical role in the commitment of myoprogenitor cells to the osteoblast lineage.[50] Our recent study also suggested that increases in activating transcription factor 4 (ATF4) levels related to the endoplasmic reticulum (ER) stress pathway brought about by Tmem119 are involved in the osteoblastic differentiation of myoblasts.[52] Moreover, our study indicated that Tmem176b and matrix metalloproteinase (MMP)-10, the expressions of which are enhanced by ALK2 signaling in myoblasts, might play some role in the differentiation of myoblasts into osteoblasts.[53,54] Sondag et al.[55] reported that osteoactivin induces the differentiation of myoblasts into osteoblasts. This approach may be a new strategy for the identification of novel bone formation factors and the development of new bone formation-stimulating agents.
Recent new evidence is accumulating in the field of mesenchymal stem cells into bone. Gsα, which stimulates cyclic adenosine monophosphate (cAMP)-dependent signaling downstream of G-protein-coupled receptors, regulates bone formation by facilitating the commitment of mesenchymal progenitors to the osteoblast lineage in association with enhanced Wnt signaling through sclerostin and dickkopf 1 (Dkk1) and by restraining the differentiation of committed osteoblasts to enable production of bone of optimal mass, quality and strength.[56] Gsα is important for the promotion of heterotopic ossification from adipose-derived mesenchymal progenitors through the membranous ossification process.[57] Zhang et al.[58] also reported that active DNA demethylation occurred during terminal specification of stem cells in an adipose-derived mesenchymal stem cell-derived osteogenic differentiation model. In that study, GADD45A played an essential role in gene-specific active DNA demethylation during osteogenic differentiation. These findings suggest that the DNA demethylation process is important for the differentiation of mesenchymal stem cells into bone.
Recipient T lymphocytes inhibit the ability of exogenously added mesenchymal stem cells to mediate bone repair through interferon-γ-induced downregulation of the runt-related transcription factor 2 (Runx2) pathway and the enhancement of TNF-α signaling in stem cells.[59] Moreover, macrophages accelerated the osteoblast differentiation of myoblastic cells induced by vascular smooth muscle cell-conditioned medium.[60]
Menin, the product of the multiple endocrine neoplasia type 1 gene, also induces the differentiation of mesenchymal cells into osteoblasts through interaction with the BMP-Runx2 pathway.[61] On the other hand, several molecules, such as Smad6 and Smad7, suppress BMP pathways.[62] Muscle-related genes are downregulated by BMPs during osteoblast commitment and differentiation. Moreover, ubiquitin conjugating enzyme 9 (Ubc9), a unique E2-SUMOylation enzyme, negatively regulates osteoblast differentiation induced by BMP via SUMOylation of Smad4 in myoblasts.[63] Zinc-finger, RAN-binding domain-containing protein 2 (ZRANB2) is also a BMP suppressor that forms a complex with Smads in the nucleus of myoblasts.[64] These findings suggest that several local negative regulators in muscle tissues might control heterotopic ossification in muscle.
Several cell populations exist in muscle. Wosczyna et al.[65] identified a tissue-resident stem/progenitor population that exhibits robust osteogenic potential and represents a major cell of origin for heterotopic ossification in the skeletal muscle interstitium.
15. Humoral factors linking muscle to bone
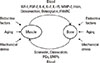
Several lines of evidence suggest certain interactions between muscle tissues and bone metabolism. Muscle tissues produce local growth factors, which have anabolic effects in bone tissues. For example, IGF-I and IGFBP-5 are secreted from muscle tissues. These findings raise the possibility that there might be some humoral factors that are produced in muscle tissues and affect bone in an anabolic fashion (Fig. 3). We hypothesized that the signal suppressed by the conversion of muscle tissues into bone might give us a clue to identify muscle-derived bone anabolic factors because those factors could be predominantly expressed in muscle tissues, compared with their expression in bone, and their systemic effects through blood could be more important than their effects in muscle tissues. We therefore selected several factors that exhibited decreased expression levels upon ALK2 (R206H) expression using comprehensive DNA microarray analysis.[66] Osteoglycin and family with sequence similarity 5, member C (FAM5C) were included in these genes.
Osteoglycin is the seventh member of the small leucine-rich proteoglycans (PGs), which may be the mechanosensitive gene that mediates an anabolic response of mechanical loading.[66] FAM5C may be related to various cellular functions as well as pathological conditions, such as atherosclerosis and inflammation.[67] The levels of osteoglycin and FAM5C as well as the effects of the conditioned medium from osteoglycin-modulated myoblastic cells were positively correlated with osteoblast phenotype and mineralization in osteoblastic cells, although those factors seemed to reduce osteoblast differentiation in osteoblasts at the early differentiation stage and in myoblasts. Moreover, osteoglycin and FAM5C proteins are detected in human serum. These findings suggest that osteoglycin and FAM5C may be crucial humoral bone anabolic factors that are produced from muscle, although clinical studies and in vivo studies using muscle-specific gene-deleted or transgenic mice are necessary.
Exercise therapy and an increase in muscle mass are considered to be very effective for an increase in BMD and a reduction in fracture risk in osteoporotic patients. However, therapy to improve these factors is clinically very difficult as the physical activity of osteoporotic patients is usually disturbed. Humoral bone anabolic factors, produced in muscle tissues, may be important as the target molecules for the treatment and prevention of osteoporosis.
There are various other factors that are produced in muscle tissues. Many of these, such as IGF-I, interleukin (IL)-15, osteonectin, MMP-2, IL-7, and FGFs, may play some roles in bone metabolism.[68] Circulating myokine, irisin, which is induced by exercise, enhances the generation of brown-like adipocytes, and systemic administration of this protein has been shown to enhance LBM. Zhang et al.[69] and Boström et al.[70] recently reported that irisin promotes osteoblast differentiation through the Wnt-β-catenin pathway and inhibits osteoclast differentiation by suppressing the receptor activator of nuclear factor-kappa B ligand (RANKL)/nuclear factor of activated T cells (NFAT)c1 pathway. Recent studies in female mice devoid of osteocalcin or osteocalcin receptor showed that they display a 10-20% decrease in muscle mass, mainly due to decreased muscle fiber diameter. Muscle fiber regeneration is compromised and the response to injury is altered in the absence of undercarboxylated osteocalcin function, suggesting that undercarboxylated osteocalcin could regulate muscle mass, function and regeneration.[71,72]
IL-6 is secreted by muscle with exercise and affects glucose and bone metabolism. Mechanically loaded myotubes secrete soluble factors other than IL-6, which affect osteoclast formation.[73] The Wnt-β-catenin signaling pathway is an important regulator of bone mass as well as muscle growth. Osteocytes are involved in the regulation of bone mass in response to mechanical stress, presumably through the production of sclerostin.[2,13] Muscle produces unknown factors that protect and preserve osteocyte viability in response to glucocorticoids.[74]
16. Myostatin
Myostatin is a member of the transforming growth factor (TGF)-β superfamily and a well-known inhibitor of skeletal muscle growth.[75] It has been suggested to have target effects on bone and tendon.[2,74] The loss of myostatin produces hyperplasia and gross hypertrophy in muscle tissues, with increases in muscular function and bone mass.[21,71,75] Conversely, mice overexpressing myostatin display muscle wasting and generalized atrophy with a cachectic phenotype. Anti-ACVR2B-Fc has been shown to increase LBM, fat metabolism and bone formation markers in postmenopausal women. However, a recent trial using this same agent in boys with muscular dystrophy was suspended because of the development of unexpected gum- and nosebleeds.[2] Moreover, a myostatin inhibitor (GDF-8 propeptide-Fc) did not alter BMD and bone strength in aged mice, although it increased muscle mass.[76] These findings suggest that pharmacological inhibition of myostatin in mice has a more pronounced effect on skeletal muscle than on bone.
17. Linkage from bone to muscle
In contrast to the links from muscle to bone, influences from bone to muscle may exist. Bone marrow mesenchymal stromal cells support osteogenesis as well as bone resorption in bone tissues. A recent study revealed that bone marrow mesenchymal stromal cells stimulate myoblast proliferation through vascular endothelial growth factor (VEGF) from mesenchymal stromal cells,[77] suggesting that bone mesenchymal cells influence muscle cells. IGF-I, MGF, myostatin, VEGF and hepatocyte growth factor (HGF) may be anabolic and metabolic factors regulating muscle mass. These factors are produced in bone cells.
Osteocytes are abundant in bone tissues and noted as endocrine cells that affect different organs, such as kidneys and parathyroid glands. A recent study showed that mechanically loaded MLO-Y4 osteocytes produce various factors, such as IGF-I, MGF, VEGF and HGF.[73] Moreover, osteocytes produce factors such as Wnt3a and prostaglandin E2 (PGE2) that support myogenesis and muscle function.[78] Gorski et al.[79] recently reported that osteocytes normally inhibit the growth and differentiation of skeletal muscle by secreting BMPs, which are modulated by circulating leptin. Therefore, osteocytes may affect muscle mass through various factors.
CONCLUSION
Muscle/bone relationships have recently been noted as a new research field related to the interactions among several organ systems. Exercise and muscle factors are clinically important for the care and treatment of osteoporosis and sarcopenia. Very few points about muscle/bone interactions are clear at the present time, but the development of an understanding of this field may lead to the development of novel drugs and biomarkers for osteoporosis and sarcopenia.

 XML Download
XML Download