

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Bone is a highly dynamic tissue that undergoes continuous remodeling, which is regulated by various factors, including cytokines/chemokines, hormones, and mechanical stimuli.[1,2] Under normal conditions, bone homeostasis is controlled by the balance between bone formation and bone resorption, processes regulated by osteoblasts and osteoclasts, respectively.[3] However, excessive bone resorption by osteoclasts compared to bone formation by osteoblasts, results in osteopenic disorders such as osteoporosis, rheumatoid arthritis, Paget's disease, and lytic bone metastases of malignancies.[2]
Osteoclast precursor cells of monocyte-macrophage lineage fuse to form tartrate-resistant acid phosphatase (TRAP)-positive multinucleated cells. The multinucleated osteoclasts reorganize the actin cytoskeleton to attach to the bone surface and to resorb the bone.[4] Osteoclast differentiation is regulated by two essential cytokines, macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-kappa B (NF-κB) ligand (RANKL). M-CSF is considered a crucial factor responsible for the survival and proliferation of osteoclast precursor cells. It also induces receptor activator of NF-κB (RANK) expression in osteoclast precursor cells to evoke efficient response to the RANKL-RANK signaling pathways. [5,6,7,8] RANKL mediates biological effects in bone through its sole receptor, RANK. Binding of RANKL to RANK receptor results in the recruitment of tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), which is involved in the activation of downstream signaling pathways, such as NF-κB, c-Jun N-terminal kinase (JNK), p38, and extracellular signal-regulated kinase (ERK) pathways.[2,9,10] In addition, RANKL activates various transcription factors such as NF-κB, microphthalmia transcription factor (MITF), c-Fos, and nuclear factor-activated T cells c1 (NFATc1), which are responsible for osteoclast differentiation.
In particular, NFATc1, a master regulator of osteoclast differentiation, regulates a number of osteoclast specific genes such as TRAP, cathepsin K, calcitonin receptor, and osteoclast-associated receptor (OSCAR) through cooperation with MITF and c-Fos.[10,11,12,13] The essential role of NFATc1 in osteoclast differentiation has been well established by several studies performed on genetically modified mutant mice. A transgenic mouse strain was generated by Winslow et al.[14] by crossing NFATc1-knockout mice with mice expressing Tie2-promoter-driven NFATc1 in order to overcome a defect in cardiac valve formation in NFATc1-knockout mice. These transgenic mice are viable and exhibit an osteopetrotic bone phenotype owing to a severe defect in the osteoclastogenesis process.[14] Another report states that osteoclast-specific conditional NFATc1-deficient mice develop osteopetrosis owing to impaired osteoclastogenesis.[15] NFATc1-deficient embryonic stem cells cannot differentiate into osteoclasts in response to RANKL. However, the ectopic expression of NFATc1 in osteoclast precursor cells induces osteoclast differentiation in these cells despite the absence of RANKL.[10] These results clearly indicate that NFATc1 is an indispensable factor for osteoclast differentiation in vitro and in vivo. Therefore, understanding the molecular mechanisms underlying the regulation of NFATc1 in osteoclasts may provide novel therapeutic strategies for bone diseases associated with excessive osteoclast differentiation and function.
NFATs
The NFAT gene family was originally identified around 25 years ago as a group of transcription factors that could bind to the interleukin 2 (IL-2) promoter in activated T cells. Since the discovery of the first NFAT protein, NFATs have been discovered to be involved in immune cell activation, heart valve formation, cardiac hypertrophy, and osteoclast development.[16,17,18,19] However, the function and regulation of NFAT proteins is best understood in T cells. The NFAT family consists of five members: NFAT1 (also known as NFATp or NFATc2), NFAT2 (also known as NFATc or NFATc1), NFAT3 (also known as NFATc4), NFAT4 (also known as NFATx or NFATc3), and NFAT5. All proteins from the NFAT family, excluding NFAT5, are regulated by the calcium signaling pathway.[20] NFAT proteins are phosphorylated at the serine residues located in the regulatory domain, and exist in an inactive form in the cytosol in resting T cells. However, the signals induce the release of intracellular Ca2+, which activates calcineurin, a ubiquitous serine-threonine phosphatase, which dephosphorylates NFAT proteins. This leads to their translocation from the cytosol to the nucleus, where they regulate the target genes.[21,22] There is a high degree of structural similarity among the different members of the NFAT family, which allows for redundancy in some NFAT functions in T cells. Knockout mice deficient in individual NFAT proteins show only mild changes in immune function; however, elimination of more than one NFAT protein in mice results in severe alterations in the immune system.[23,24,25,26] These results indicate some degree of redundancy among NFAT proteins in T cells.
Transcriptional regulation of NFATc1 in osteoclasts
NFATc1 expression is induced during osteoclast differentiation. Several transcription factors have been found to bind to NFATc1 promoter during osteoclastogenesis.
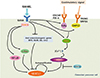
c-Fos, a member of the activator protein-1 (AP-1) family of transcription factors, is induced at an early stage during osteoclast differentiation. c-Fos-knockout mice develop osteopetrosis owing to defects in osteoclast lineage commitment.[27,28] In addition to osteoclast differentiation, the induction of NFATc1 mRNA by RANKL is also abrogated in c-Fos-deficient cells. These defects were corrected by overexpression of NFATc1 in c-Fos-deficient cells in vitro.[10,29] Studies have also reported that c-Fos is recruited to the NFATc1 promoter at an early stage of osteoclast differentiation.[30,31] These results suggest that c-Fos is an indispensable factor involved in early induction of NFATc1 for osteoclast differentiation (Fig. 1).
RANKL also rapidly stimulates the activation of classical or alternative NF-κB pathway in osteoclast precursor cells.[31] The NF-κB family, a group of dimeric transcription factors, consists of five members: cRel, RelA (p65), RelB, NF-κB1 (p50), and NF-κB2 (p52). In the classical pathway, inhibitor of κB (IκB) kinase (IKK) complex phosphorylates and degrades IκB. Subsequently, the proteasomal degradation of IκB induces activation of the p50/RelA complex.[32,33] In the alternative pathway, NF-κB-inducing kinase (NIK) and IKKα phosphorylate and process p100 to generate p52 by proteasomes, resulting in the activation of the p52/RelB complex.[32,33,34,35] The important role played by NF-κB proteins in osteoclastogenesis has been verified by the severe osteopetrotic phenotype observed in p50 and p52 double deficient mice.[36,37] Recently, it has been reported that dehydroxymethylepoxyquinomicin (DHMEQ), an NF-κB inhibitor, attenuates RANKL-induced osteoclastogenesis through down-regulation of NFATc1.[38] Consistent with these results, ChIP experiments have demonstrated that the NF-κB components p50 and p65 are recruited to the NFATc1 promoter 1 hr after RANKL stimulation.[30] Although it is unclear whether the classical or alternative NF-κB pathway is dominant in osteoclast differentiation, it is certain that the NF-κB components p50 and p65 are important for the initial induction of NFATc1 during RANKL-induced osteoclastogenesis (Fig. 1).
The NFAT protein family plays a redundant role in the immune system. NFATc1 and NFATc2, closely related due to the similarity in their DNA binding domains, are expressed in osteoclasts. However, NFATc1-deficient mice exhibit a severe osteopetrotic phenotype, while NFATc2-deficient mice show normal skeletal development.[15,30,39] To investigate the unexpected non-redundant role played by NFATc1 in the skeletal system, Asagiri et al.[30] ectopically expressed NFATc1 and NFATc2 in NFATc1-deficient osteoclast precursor cells. The defect in osteoclast formation by NFATc1-deficient osteoclast precursor cells is recovered by overexpression of NFATc1 and NFATc2. They also established that FK506, an inhibitor of NFAT activity, suppresses mRNA expression of NFATc1 but not of NFATc2.[30] ChIP experiments revealed that NFATc1 is consistently recruited to the NFATc1 promoter, but not the NFATc2 promoter, during the terminal differentiation of osteoclast.[30] Therefore, NFATc1 is suggested to be a unique NFAT protein, which is regulated at the transcriptional level through an autoregulatory loop during osteoclast differentiation. Interestingly, NFAT-binding sites exist in NFATc2 as well as NFATc1 promoters. During osteoclast differentiation, transcriptional coactivators with histone acetylase activity, including the cyclic adenosine 3',5'-monophosphate (cAMP) response element-binding protein (CREB)-binding protein (CBP) and p300/CBP-associated factor (PCAF), are recruited to NFATc1 promoters but not NFATc2 promoters.[30] In addition, histone deacetylase 1 (HDAC1) gradually dissociates from NFATc1 promoters as the osteoclasts differentiate.[30] Therefore, the exclusive autoamplification of NFATc1 in osteoclasts is supported by epigenetic regulation of the NFATc1 promoters. In conclusion, the induction of high levels of NFATc1 transcriptional factor by RANKL induces the self-sustaining positive autoregulatory system to maintain sufficient NFATc1 expression and osteoclast differentiation (Fig. 1).
As NFATc1 is a master transcriptional factor for osteoclast differentiation, our research, as well as that of others, was focused on finding the negative regulators of NFATc1 in osteoclasts. Our research has indicated that RANKL attenuates expression of the inhibitors of differentiation (Ids), V-maf musculoaponeurotic fibrosarcoma oncogene homolog B (MafB), and LIM homeobox 2 (Lhx2), which act as negative regulators of osteoclastogenesis by downregulating the expression of NFATc1 (Fig. 1).[40,41,42] We have also found that protein inhibitor of activated STAT3 (PIAS3) inhibits RANKL-mediated transcription of NFATc1 (Fig. 1).[43] Interferon regulatory factor-8 (IRF-8) and B-cell CLL/lymphoma 6 (Bcl-6) have also been identified as negative regulators of NFATc1.[44,45] RANKL inhibits the expression IRF-8 during osteoclastogenesis, and the overexpression of IRF-8 blocks RANKL-induced osteoclast differentiation.[44] Bcl-6 inhibits osteoclastogenesis by suppressing the expression of NFATc1 and other genes (Fig. 1).[45] In addition, the deletion of Lhx2, IRF-8, and Bcl-6 in mice causes severe osteoporosis.[42,44,45] These results demonstrate the importance of multi-pathway regulation of NFATc1 for maintenance of normal bone homeostasis.
Epigenetic regulation of NFATc1 in osteoclasts
Epigenetics is defined as heritable change in the function of genetic elements without changes in the DNA sequence.[46] There are three classes of epigenetic markers, DNA methylation, histone modification, and noncoding RNAs. These play an important role in the determination of cell fate.[47]
Among the epigenetic modification methods, DNA methylation is understood the best. Generally, hypermethylation of CpG-rich regions in gene promoters blocks gene expression, while hypomethylation stimulates gene expression.[47,48] Yasui et al.[49] recently used ChIP sequencing technology to demonstrate that histone H3 lysine 4 (H3K4me3) is present in the NFATc1 gene in osteoclast precursor cells, but is markedly reduced in mature osteoclasts. During osteoclastogenesis, expression and recruitment of jumonji domain-containing protein 3 (Jmjd3), a H3K27 demethylase, around the transcription start site of NFATc1 is induced. In addition, the down-regulation of Jmjd3 by using short hairpin RNA, inhibits demethylation of H3K27me3 at the transcription start site of NFATc1; thereby suppressing RANKL-induced osteoclast differentiation. These results raise the possibility that epigenetic regulation of NFATc1 by the mechanism underlining methylation or demethylation is essential for RANKL-induced osteoclast differentiation.
Post-translational modification such as ubiquitination and acetylation are important mechanisms of gene regulation.[50] Our previous studies have indicated that the stability of NFATc1 proteins is controlled by ubiquitination and acetylation during osteoclast differentiation (Fig. 2).[51]
NFATc1 proteins are downregulated by M-CSF during late stage osteoclastogenesis, and subsequently degraded through the ubiquitin-proteasome pathway in the cytoplasm. In addition, NFATc1 interacts endogenously with c-Src, c-Cbl, and Cbl-b in the osteoclasts. Overexpression of c-Src induces down-regulation of NFATc1, and depletion of the Cbl proteins blocks NFATc1 degradation during late stage osteoclastogenesis. These results suggest that M-CSF induces NFATc1 ubiquitination and degradation via Cbl proteins in a Src kinase dependent method during late stage osteoclastogenesis.[51] Therefore, our previous research data suggest a negative regulatory mechanism through ubiquitination of NFATc1, mediated by M-CSF signaling pathway, in osteoclast differentiation (Fig. 2).
RANKL increases the stability of the NFATc1 proteins through induction of NFATc1 acetylation. We demonstrated that RANKL could induce the accumulation of NFATc1, which is regulated by acetylation of the NFATc1 protein via a transcription-independent mechanism. NFATc1 acetylation is mediated by histone acetyltransferase (HAT) activity of PCAF through physical interaction with NFATc1.[52] Because acetylation of many proteins is a reversible post-translational modification, we have identified which HDAC can inhibit lysine acetylation by removal of the acetyl group from the NFATc1. Particularly, HDAC5 greatly inhibits NFATc1 acetylation, among the HDAC protein group. In addition, HDAC5 reduces the stability and transactivation of NFATc1, thereby attenuating RANKL-induced osteoclast differentiation (Fig. 2).[52] Therefore, our study proves that M-CSF and RANKL, two essential cytokines, induce ubiquitination and acetylation of NFATc1 respectively, to control osteoclast differentiation, by modulating NFATc1 stability and activity.
miRNAs are short non-coding RNAs, approximately 22 nucleotides in length, that regulate diverse biological processes, including proliferation, apoptosis, and differentiation, through post-transcriptional regulation. Generally, miRNAs act as direct negative regulators of gene expression that bind to the specific sequence at the 3' untranslated region (UTR) of the target mRNA.[53,54] The miRNA biogenesis pathway has multiple steps: transcription, pri-miRNA processing, transport to the cytoplasm, precursor miRNA processing, strand selection, target transcription, and fate transcription.[55,56] Very little is known about the role of miRNAs during osteoclast differentiation. Recent reports have revealed the unprecedented regulation of NFATc1 by miRNA in osteoclasts. Lee et al.[57] have shown that miRNA-124 regulates osteoclastogenesis by suppressing NFATc1 expression (Fig. 2). With emerging evidence suggesting the involvement of several miRNA in osteoclastogenesis, additional studies will be required to find an miRNA that can directly regulate NFATc1 expression in osteoclasts.
Regulation of NFATc1 by Ca2+ signaling and costimulatory signaling in osteoclasts
It has been substantially demonstrated that NFAT proteins are primarily regulated by calcium and calcineurin. RANKL activates phospholipase C-γ (PLC-γ), which hydrolyses phosphatidylinositol-4,5-bisphosphate to generate inositol-1,4,5-trisphosphate (InsP3) and diacylglycerol. InsP3 induces the release of calcium from the endoplasmic reticulum Ca2+ stores. Calcium binds to calmodulin, which in turn activates the calmodulin-dependent phosphatase calcineurin. Dephosphorylation of serine residues in NFATc1 by activated calcineurin leads to nuclear translocation, and activation of NFATc1 proteins. Targeted deletion of PLC-γ2 in mice results in an osteopetrotic phenotype, and the osteoclast precursor cells lacking PLC-γ2 do not sufficiently induce NFATc1 expression even in the presence of RANKL.[58] Well known inhibitors of calcineurin, such as FK506 and cyclosporine A, strongly inhibit RANKL-induced osteoclast differentiation by blocking NFATc1 translocation into the nucleus in vitro and in vivo. In addition, the Ca2+ chelator BAPTA-AM suppresses osteoclast differentiation mediated by RANKL through the inhibition of nuclear localization of NFATc1.[10] These results suggest that the activation of calcium-calcineurin pathway plays a crucial role in the regulation of NFATc1 during osteoclastogenesis.
Several kinases such as glycogen synthase kinase 3 (GSK3), casein kinase 1 (CK1), p38, and JNK phosphorylate the various serine-rich motifs in NFAT proteins, thereby preventing translocation into the nucleus or promoting nuclear export of NFAT proteins.[59,60,61,62,63] A recent study has reported that the ectopic expression of the constitutively active GSK-3β (GSK3β-S9A) mutant in osteoclast precursor cells inhibits RANKL-mediated NFATc1 expression and Ca2+ oscillations. Furthermore, Jang et al.[64] have also shown a significant defect in NFATc1 expression and nuclear localization in osteoclast precursor cells isolated from transgenic mice expressing GSK3β-S9A mutant. These findings indicate that GSK3β inactivation by RANKL is important for the expression and activation of NFATc1 in osteoclasts (Fig. 1).
Since RANK receptor does not seem to directly initiate Ca2+ signaling, it has been studied for its role as an immunoreceptor alongside other receptors such as OSCAR, triggering receptor expressed in myeloid cells-2 (TREM-2), paired immunoglobulin-like receptor-A (PIR-A), and signal-regulatory protein β1 (SIRPβ1) in osteoclasts that are involved in calcium signaling in immune cells.[65] The association of immunoreceptors with immunoreceptor tyrosine-based activation motif (ITAM)-harboring adaptors has been observed in osteoclast precursor cells: OSCAR and PIR-A are associated with Fc receptor common γ subunit (FcRγ) and TREM-2 and SIRPβ1 are associated with DNAX-activating protein 12 (DAP12). The association of an immunoreceptor with the ITAM-harboring adapters, FcRγ and DAP12, stimulates calcium signaling. Although DAP12-deficient mice exhibit only mild osteopetrosis, FcRγ and DAP12 double knockout mice (DAP12-/- FcRγ-/-) develop severe osteopetrosis due to defects in osteoclast differentiation.[65,66,67] Interestingly, the defect in osteoclast differentiation in DAP12-/- FcRγ-/- cells is fixed by the overexpression of DAP12, but not DAP12Y65F, which has a mutation in ITAM, suggesting that the ITAM-mediated signaling pathway is indispensable for osteoclastogenesis.[65] As expected, Ca2+ oscillation and the induction of NFATc1 were also suppressed in DAP12-/- FcRγ-/- cells, recovered by the overexpression of NFATc1, but not c-Fos or TRAF6.[65] These results suggest that the induction and activation of NFATc1 is regulated by calcium signaling pathway, mediated by ITAM-harboring adapters, FcRγ and DAP12, in the osteoclasts (Fig. 1).
The stimulation of ITAM-associated receptors alone, without RANKL, cannot induce osteoclast differentiation, although ITAM-associated receptors stimulate Ca2+-NFATc1 pathway through ITAM-harboring adaptors during osteoclastogenesis. Therefore, ITAM-mediated signals may provide costimulatory signals to maximize the induction of NFATc1 for RANK.[9]

 XML Download
XML Download