

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Bone fracture repair involves the coordinated expression of growth factors and signaling molecules that regulate the ordered development of the fracture callus. Following the initial inflammatory response, fracture callus soft tissues proliferate and ossify to bridge the injury with woven bone that is eventually remodeled to cortical bone, a sequence of healing stages that facilitates the evaluation of bone repair and the efficacy of therapeutic approaches.[1] Therapies that improve bone healing impaired by age or disease are highly desirable, and experimental efforts have attempted to promote different aspects of bone repair to improve the healing process. Therapeutic studies have generally identified novel approaches to enhance bone formation during tissue development and repair and applied them to the fracture tissues at a critical point in healing when they might be beneficial to repair. Several gene therapy and protein therapy approaches have successfully augmented growth factor and signaling molecule expression in bone fractures to improve impaired healing.[2] The application of therapy to enhance an early phase of tissue repair would seem highly beneficial to impaired bone healing.
The inducible inflammatory mediator cyclo-oxygenase-2 (Cox-2) has a critical role regulating tissue homeostasis. Cox-2 expression is up-regulated well into the endochondral stage of fracture repair.[3,4] The expression of Cox-2 has been demonstrated to be necessary for bone repair, clinically through impaired bone fracture repair in rheumatoid arthritis patients under treatment with the non-steroidal anti-inflammatory inhibitor drugs, and experimentally, in mice deficient in Cox-2 expression that exhibit impaired healing of endochondral bone fractures.[5,6,7]
Cox-2 functions through the production of prostaglandins (PGs), especially PG E2 (PGE2), which modulates Cox-2 effects through four PGE2 receptors (PTGER). The PTGERs are expressed on a wide variety of cells and act through cyclic adenosine monophosphate (cAMP) and inositol triphosphate (IP3) intracellular signaling pathways to differentially regulate the cell response to PGE2 and to regulate tissue development and repair.[8] PTGER effects on bone repair are diverse; PTGER1 expression inhibits bone repair, [9] but agonists for PTGER2 and PTGER4 stimulate bone formation, and promote endochondral bone repair.[10,11] These observations indicate that Cox-2 production of PGE2 initiates a complex regulation of different aspects of bone repair.
We have previously demonstrated that Cox-2 gene therapy could indeed improve endochondral bone fracture repair.[12] However, the therapeutic effect was unexpectedly not seen until the endochondral stage of bone fracture repair, during fracture chondrogenesis, when the callus cartilage is being remodeled to bone and well after the inflammatory stage has subsided, during which time Cox-2 expression has been observed to initiate fracture healing.[13] We speculate that while Cox-2 functions might also mediate some aspects of post-inflammatory bone healing during endochondral bone repair. Because Cox-2 also mediates angiogenesis and tissue remodeling during certain pathogenic conditions, such as metastasis,[14,15] and because angiogenesis coincides with the ossification of fracture cartilage,[16] we hypothesized that Cox-2 gene expression might also regulate angiogenesis and extracellular matrix remodeling of the fracture callus cartilage to promote bony union of the fracture during endochondral bone repair.
To elucidate the molecular pathways regulating Cox-2 promotion of bony union during endochondral bone repair, we employed an in vivo gene transfer approach to express a human Cox-2 (hCox-2) transgene in murine fracture tissues, and characterized the response to hCox-2 over-expression through a microarray analysis of global gene expression at this stage of fracture healing, i.e., at 10 days post-fracture.
METHODS
Closed femoral fractures were produced in 12-week-old C57BL/6 male mice by the three-point bending technique.[17] The bone fracture model was examined because Cox-2 expression is normally limited to injured tissues. Male mice were used because Cox-2 deficiency has been observed to affect males more than females, suggesting that Cox-2 gene therapy might have a greater effect on fracture repair in males.[18]
An in vivo gene transfer approach that expressed a modified hCox-2 transgene in fracture tissues had previously been highly effective in promoting bony union of fracture gaps in a rodent femur fracture model.[12,19] The use of hCox-2 gene is to allow us to distinguish transgene expression from endogenous Cox-2 gene expression by real-time reverse transcription-polymerase chain reaction (RT-PCR) with species-specific Cox-2 primers. The hCox-2 gene is modified by removing most of the 3'untranslated region (3'UTR) sequence and replacing the endogenous Kozak sequence with an enhanced Kozak sequence to increase the stability and translation efficiency of the mRNA.[12] The current study has confirmed significant expression of the hCox-2 transgene in the Cox-2-treated fracture tissues used in this study by microarray and real-time RT-PCR (data not shown).
On the day after fracture, a murine leukemia virus (MLV)-hCox-2 expressing vector was delivered to the lateral and medial aspects of the periosteum at the fracture site.[12] Three fracture samples injected with the hCox-2 transgene were compared with three fracture samples injected with the β-galactosidase control transgene. Fracture tissues were harvested at 10 days post-fracture, a time chosen for microarray analysis because Cox-2 gene expression is elevated at this time,[4,20] but most importantly because it is just prior to Cox-2 promotion of endochondral bony union[12] when we should expect the genes mediating this function to be expressed in response to Cox-2. To evaluate the complete repertoire of genes that might coordinate bony union among the various fracture tissues, the entire fracture callus was harvested, separated from the femoral epiphyses, pulverized in Trizol (Life Technologies, Grand Island, NY, USA) and the total RNA isolated. The quality of RNA was confirmed by Bioanalyzer analysis (Agilent Technologies, Santa. Clara, CA, USA). All animal procedures were approved by the local Institutional Animal Care and Use Committee.
To confirm gene expression of the hCox-2 transgene in the fracture tissues, the RNA from these fracture samples that underwent microarray analysis was reversed transcribed to cDNA, and hCox-2 transgene expression was confirmed by real-time RT-PCR of the fracture callus RNA with the SYBR green master mix (Applied Biosystems, Foster City, CA, USA) in an Opticon Chromo4™ system (Bio-Rad, Hercules, CA, USA) with Opticon Monitor 3.1 software (Bio-Rad), using hCox-2 gene-specific primers (Integrated DNA Technologies, Coralville, IA, USA) (Table 1). The real-time RT-PCR approach was also used to determine the expression of the four PTGER genes in response to hCox-2 transgene expression relative to the expression of the housekeeping gene, peptidylprolyl isomerase A (PPIA, cyclophilin A). Statistical analysis was performed by t-test.
The Affymetrix Mouse Gene 1.0 ST array was used for hybridization (Affymetrix, Santa Clara, CA, USA). This array represents 28,853 genes, with each target gene queried by a median number of 27 individual 25-mer oligonucleotide probe sequences. Image analysis and signal normalization was performed at the University of California at Irvine using the "Probe Logarithmic Error Intensity Estimate" (PLIER) algorithm, a normalization that improves the signal for genes with smaller changes in expression.
The gene expression and literature search software "Pathway Studio" (version 9, Elsevier, Philadelphia, PA, USA) were used for the analysis of gene regulation in response to Cox-2 transgene expression. Genes with significant changes in expression were furthered analyzed by "gene set enrichment analysis" (GSEA)[21] and were classified into the "Gene Ontology" (GO) "Biological Function" categories (statistically significant at P<0.05 by Kolnogorov-Smirnov test, 400 gene permutations). For this analysis, the entire repertoire of genes that displayed significant differences in expression in the initial PLIER normalization was examined. GSEA provided the enrichment score (ES) by classifying the genes with significant changes in expression into the GO Biological Function category relative to the total number of genes from that GO category on the chip. "Leading Edge Analysis" further identified the gene sets with the greatest representation in the repertoire of expressed genes. Those gene sets predicted to mediate the greatest regulation of Cox-2 transgene effects were then identified. Accordingly, the "leading edge" of the GSEA of the ES of these gene sets was identified as those ranked categories preceding maximum positive ES for positively enriched gene sets, and those sets following the minimum negative ES for negatively enriched gene sets. The positive and negative "leading edge" GO gene sets were then ranked by the normalized ES (NES) and presented with their median change in expression.
Further analysis examined gene expression among the individual genes with the greatest changes in magnitude of expression. This analysis of the microarray results was performed using Biometric Research Branch (BRB)-ArrayTools Version 4.1.0 (Biometric Research Branch, National Cancer Institute, Rockville, MD, USA). An arbitrary threshold of a 1.5-fold positive or negative change in expression was used to identify the affected genes. A gene was excluded when less than 20% of its expression data displayed the minimum 1.5-fold change in either direction from its median expression value. We identified genes that were differentially expressed between the two classes using a random-variance t-test. Genes were considered statistically significant if the P-value of the log-ratio variation was less than 0.01 (P<0.01), but were excluded if the minimum value of the spot intensity was less than a threshold of 10.
To confirm the microarray gene expression profile results, the expression of several key inflammatory and hematopoietic genes (Table 2) was also determined by real-time RT-PCR, using the same RNA samples that were used in the microarray analysis. The fold-increase in expression was determined by the 2-ΔΔCt method. The expression of these inflammatory, hematopoietic, and remodeling genes determined by real-time RT-PCR was then correlated with that determined by the microarray.
RESULTS
The expression of the hCox-2 transgene in these fracture callus tissues was verified by real-time RT-PCR with hCox-2-specific primers to be at least 7 cycles, or more than 128-fold, above background levels, confirming that the in vivo gene transfer approach did indeed augment Cox-2 gene expression in the fracture tissues.[12,19] We previously established that Cox-2 gene therapy augmented PGE2 expression in the rodent fracture model,[12] confirming that the hCox-2 product was functional. That study also established that hCox-2 transgene expression did not alter the expression of the endogenous murine Cox-2 gene (data not shown), and suggested that there was no feedback regulation of the endogenous murine Cox-2 gene in response to the hCox-2 gene therapy.[12]
An initial examination identified individual genes that displayed significant changes in expression. Of the 28,853 genes represented on the chip, 1,031 individual genes displayed significant differences (P<0.01) in expression between the Cox-2 and β-galactosidase transgene-injected fractures. Of these genes, 433 were up-regulated and 598 were down-regulated, indicating that Cox-2 regulated many genes, probably functioning in several different pathways.
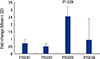
Only one of the PTGER exhibited significant changes in expression in response to Cox-2 transgene expression. PTGER3 was up-regulated 1.4-fold by microarray analysis, and confirmed by real-time RT-PCR to be up-regulated 2.6-fold (P<0.04) at 10 days post-fracture (Fig. 1). None of the other PTGER exhibited significant changes in gene expression at 10 days post-fracture between the Cox-2 and control gene samples, either by microarray or by real-time RT-PCR analysis. Real-time RT-PCR confirmed that PTGER3 expression was up-regulated 8-fold (P<0.04) in response to Cox-2 transgene expression at 5 days post-fracture, indicating that its expression might have also been induced during inflammation in bone repair but persisted to at least 10 days post-fracture, at which time when the benefits of Cox-2 gene therapy on bony bridging were seen in fracture repair.[12]
GSEA (Table 3)
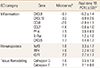
A "leading edge" analysis of the GSEA classified genes with different magnitudes of changes in expression into various functional "GO" categories, providing a more comprehensive method for identifying molecular pathways associated with transgene expression. By this approach, the gene sets containing regulators of cell proliferation were very highly represented. These gene sets included several categories of genes that have functions in the regulation of mitosis and cell proliferation.
Among the genes with significant changes in expression, the growth factors traditionally assigned angiogenic functions that would be expected to mediate this stage of bone fracture repair were notably absent. Specifically, the members of the vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) gene families were absent.
Several inflammatory genes, notably the inflammatory chemokines, were reduced in the "leading edge" analysis, suggesting that inflammation was negatively regulated. As a group, the genes with inflammatory functions were some of the most frequently represented in the entire microarray analysis in terms of numbers of genes, but also some of the most down-regulated in terms of expression.
In contrast, the hematopoietic and erythropoietic genes were the most abundant gene sets in the GSEA, with several GO categories represented. This observation indicates that a significant consequence of Cox-2 transgene expression at this stage of fracture repair involved blood cell proliferation and development. Many of these gene sets also displayed the greatest increases in expression among all gene sets, while several others displayed smaller but still significant changes in expression.
Individual Gene Analysis (Table 4)
Further analysis examined the individual genes with the greatest changes in expression. Among the genes that exhibited at least 1.5-fold changes in expression in the fracture tissues in response to Cox-2 transgene expression, 172 genes were up-regulated and 99 genes were down-regulated more than 1.5-fold (P<0.01).
The inflammatory genes were the most dramatically down-regulated in the individual gene analysis (Table 4). The 1.5-fold down-regulated genes included several chemokines, such as the monocyte chemoattractants chemokine ligand (CCL)-7, CCL-8, CXC chemokine ligand (CXCL)-9 and CXCL-10. The sole chemokine receptor up-regulated 1.5-fold was CXC chemokine receptor (CXCR)-5. In addition, platelet factor-4 (PF-4) and interleukin-7 alpha (IL-7α) receptor (IL-7αr) were among the few inflammatory regulators with expression up-regulated 1.5-fold. Thus, the inflammatory genes were not only well-represented in the microarray analysis in terms of numbers of individual genes, but also displayed some of the greatest decreases in expression in response to hCox-2 fracture therapy.
As in the GSEA, the hematopoietic and erythropoietic genes were also well-represented among the genes that were up-regulated more than 1.5-fold (Table 4). Many of the most up-regulated gene products are expressed during blood cell development. It also included a repertoire of transcription factors related to these pathways. For example, Kruppel-like factor 1 (KLF1, 2.2-fold) and IKAROS family zinc finger 3 (Ikzf3, 1.8-fold) were significantly up-regulated in this analysis, and these transcription factors regulate erythropoiesis, and lymphocyte development, respectively.[22,23,24] Interferon regulatory factor-4 (IRF-4), which functions in the development of T-lymphocytes, was up-regulated 1.8-fold.
The proliferation regulating genes were not nearly as well represented among the 1.5-fold regulated genes as they were in the GSEA. There were selected genes implicated in cell cycle progression, such as cyclin F and yippee-like-4 (Ypel-4),[25] but it appears that the individual cell cycle regulators were not highly up-regulated in expression.
The genes regulating the extracellular matrix organization and cell adherence were less abundant than in the control fracture tissues. However, Cox-2 transgene expression increased the expression of osteoclast-associated tissue remodeling gene expression. The expression of cathepsin E, cathepsin G and carbonic anhydrases 1 and 2 was up-regulated more than 1.5-fold. On the other hand, only two of the matrix metalloproteinase (mmp) genes, mmp-3 and mmp-12, exhibited increases in expression, and this up-regulation was below the 1.5-fold threshold.
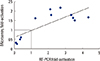
To confirm the microarray gene profiling results, we also performed real-time RT-PCR analysis of expression of selected genes of several GO categories using the same RNA samples that were used in the microarray analysis (Table 2). Fig. 2 shows a strong positive correlation between the relative expression levels of several pro-inflammatory cytokine genes (PF-4, IL-7αr and the different chemokines), hematopoietic and erythropoietic genes (KLF1, Ikzf3, IRF-4), and extracellular proteases (cathepsin E and cathepsin G) determined by microarray and those determined real-time RT-PCR.
DISCUSSION
Initially, a GSEA organized according to the gene GO "Biological Function" category examined the changes in gene expression in response to Cox-2 transgene expression. However, this analysis immediately suggested that Cox-2 inhibits inflammation but promotes blood cell development at this stage of fracture repair (Table 3).
In a further analysis the expression of individual 1.5-fold regulated genes, a limited number of pro-inflammatory genes displayed increased expression in response to Cox-2 transgene expression, notably PF-4 (Table 4). The chemokine receptor CXCR-5, a regulator of B cell trafficking[26] was up-regulated, as was IL-7αr, a regulator of T cell development.[27] However, most inflammatory genes were down-regulated. The monocyte attractant chemokines CXCL-9 and CXCL-10[28,29] were more than 2-fold down-regulated, and were among the most down-regulated genes on the microarray. Other inflammatory chemokines were also down-regulated more than 1.5-fold, notably the monocyte trafficking chemokines CCL-7 and CCL-8.[30] These results were intriguing and somewhat unexpected, since Cox-2 has been traditionally assigned pro-inflammatory functions, at least during the initial stages of tissue repair. These findings suggest that Cox-2-derived PG products can promote bony union by inhibiting inflammatory gene expression, and raises the interesting possibility that the inflammatory response must subside before bony union can occur.
In addition to several up-regulated antigen genes associated with hematopoietic development observed in the GSEA, intracellular signaling pathways that have been described in hematopoietic cell development were up-regulated (Table 4), including pathways for the genes pyruvate kinase, liver and red blood cell (Pklr, liver and red blood cell), the Bruton gammaglobulinemia tyrosine kinase (Btk) and CXCR-5. In particular, the genes for IRF-4, a lymphocyte regulator, and the transcription factor KLF1, a regulator of stem cell contributions to erythropoiesis,[24] exhibited significant increases in expression of 1.8-fold and 2.2-fold, respectively. These results correlated well between the real-time microarray and RT-PCR approaches.
It is possible that the erythropoietic and hematopoietic gene expression was actually secondary to blood vessel development from the expression of angiogenic growth factors, such as VEGF, prior to the healing time that we examined. However, because 1) hematopoietic stem cell genes are induced during fracture repair,[31] 2) hematopoietic stem cell expansion has been demonstrated to be dependent on PGE2 production,[32] and 3) the PGs promote diverse aspects of erythropoietic and hematopoietic progenitor cell proliferation, survival and development,[33] it is plausible that Cox-2 transgene expression at 10 days post-fracture enhances bony union through hematopoiesis. The absence of changes in CXCL-12 and CXCR-4 expression in this analysis also suggests that angiogenic effects of PGE2 are not mediated through endothelial cells,[34] although our gene therapy approach was effective in promoting hematopoiesis by targeting Cox-2 transgene expression to periosteal cells.
The promotion of hematopoiesis might have suppressed the inflammatory reaction through the production of myeloid derived suppressor cells (MDSCs). MDSCs can be induced by PGE2[35] and can inhibit inflammatory responses through suppressive functions on T cells in adaptive immunity and macrophages in innate immunity,[36] characteristics that might explain the reduction of inflammatory gene expression in response to a pro-inflammatory mediator such as PGE2.
The results of the GSEA were also surprising because the genes traditionally assigned angiogenic roles that we expected to be expressed were not represented. In this respect, our microarray analysis agrees with that of Hadjiargyrou et al.[3] On the other hand, the members of the FGF axis were expressed in a microarray analysis of early fracture repair.[4] Other fracture repair studies have described the response of healing to VEGF therapy in the rabbit radius[37] and in the multiple tibial fracture model.[19] In the latter case, increased expression of VEGF genes was observed, but slightly after our harvest time of 10 days post-fracture. Additionally, the multiple fracture approach used in this study might have exposed more marrow cell targets than the periosteal cells targeted in this study and promoted angiogenic growth factor expression.[8]
The expression of the Cox-2 transgene has been established to up-regulate PGE2 production,[12] whose effects are mediated through the 4 PTGER receptors. PTGER3 expression was up-regulated in response to Cox-2 transgene expression by 1.4-fold in the microarray analysis, and confirmed as 2.6-fold up-regulated by real-time RT-PCR analysis, suggesting that PTGER3 was important in mediating PGE2 effects in fracture repair at this time (Fig. 1). This receptor can generate different responses to PGE2 signaling through three different isoforms of its receptor.[38] PTGER3 has been associated with angiogenesis in acute and tumor-related chronic inflammation[39,40,41] and with VEGF functions in wound healing angiogenesis,[42] although the regulation described in those studies was post-transcriptional and would not have been observed by a microarray approach.
There were remarkably few bone formation genes represented in the microarray analysis by the GSEA analysis (Table 3) or the individual gene analysis (Table 4), despite observations that PGE2 can regulate bone morphogenetic protein-2 (BMP-2) expression.[43] Sex determining region Y-box 9 (Sox-9), an important regulator of chondrocyte commitment, was down-regulated, but only by 1.4-fold. Nevertheless, this result is consistent with our previous proposal that Cox-2 gene therapy enhances bony union by suppressing cartilage formation and promoting cartilage degradation.[37]
The sets of genes regulating osteoclast-related genes were enriched and displayed increased expression (Table 4), consistent with a Cox-2-mediated increase in bone resorption during healing. The up-regulation of the osteoclast-related genes cathepsin E, cathepsin G and the carbonic anhydrases 1 and 2 support this argument. However, the absence of changes in expression among the mmp genes in this analysis was unexpected, as mmp-9 is an established regulator of fracture callus remodeling,[44] and PTGER3 up-regulates the expression of both mmp-9 and VEGF.[45]
Because this study was designed to identify possible regulatory pathways that mediate Cox-2 functions during endochondral bone repair, 1) Cox-2 expression was enhanced by gene therapy, 2) gene expression was examined at a single time, and 3) the samples examined were limited in number. The gene expression results should therefore be further characterized at other times and with additional fracture samples. Additionally, although the high Cox-2 gene expression in fracture tissues treated with the Cox-2 in vivo gene transfer approach was certainly not physiological, Cox-2 gene therapy did promote fracture healing in the fracture model, and gene expression identified by this approach might identify molecular pathways of fracture repair for further investigation.
A model for Cox-2 gene therapy for endochondral bone fracture repair is presented in Fig. 3. In this model, endogenous Cox-2 normally promotes inflammation in early bone healing, but inhibits inflammation and enhances hematopoiesis later in healing. The connection to fracture angiogenesis illustrated by dotted arrows is inferred from another study.[19] We conclude that the expression of Cox-2 gene therapy promotes bony union by up-regulating erythropoiesis- and hematopoiesis-related gene expression, but also surprisingly inhibits inflammation.
In conclusion, Cox-2 transgene expression promoted the expression of genes regulating the proliferation and development of hematopoietic blood cell precursors, but surprisingly did not up-regulate the expression of angiogenic growth factor genes. The inflammatory genes were down-regulated, which was unexpected, given the proinflammatory role of PGs. Cox-2 gene therapy could promote bony union through hematopoietic precursor proliferation and development during endochondral bone repair.




 XML Download
XML Download