

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Skeletal growth involves dynamic changes in bone shape and size through a process termed bone modeling. Once the adult skeleton has matured, homeostasis is maintained by the bone remodeling process.[1] Bone is remodeled through the complex multicellular activities in an anatomical structure called the basic multicellular unit (BMU) involving various types of cells, although mainly osteoclasts and osteoblasts.[2] The cellular activities in each BMU proceed through four distinct phases: activation of the quiescent bone surface, recruitment of osteoclasts and bone resorption, coupling from resorption to formation or a reversal from catabolism to anabolism, and recruitment of osteoblasts and bone formation. The cellular and molecular mechanisms by which a new BMU site is determined, the catabolic activities are appropriately followed by bone anabolism and all of these activities cease remain mostly enigma.
The most dynamic and dramatic event during bone remodeling is the phase conversion from catabolism to anabolism, namely the coupling process of bone formation to the preceding resorption activity. Historically, growth factors, such as transforming growth factor-β (TGF-β) and insulin-like growth factor (IGF), that are stored abundantly in the bone matrix and released and activated as a result of bone resorption, have been implicated as the signals that link bone resorption to formation, and recent mouse genetics studies have added some experimental support for this longstanding concept.[3,4] Emerging evidence points to the involvement of factors secreted or presented by osteoclasts themselves in the coupling process.[5]
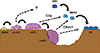
Here we review the findings on osteoclast products that may function as potential coupling factors, including two secretory products that were recently identified in our laboratory (Fig. 1).[6,7]
EphrinB2 (EFNB2)
EFNB2 was first identified as a target of nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1), a master transcription factor that drives osteoclast differentiation, and its expression was shown to be induced during osteoclastogenesis.[8] Osteoblasts express multiple ephrin (EPH) ligands and receptors, among which EPH receptor B4 (EphB4) seems to function as a receptor that transduces "reverse" signaling from osteoclastic EFNB2, since knockdown of EphB4 resulted in a decrease in alkaline phosphatase (ALP) activity.[8] When EphB4 was overexpressed in osteoblastic cells using a type I collagen promoter in vivo, increased bone formation and decreased osteoclast number were observed, with a net increase in bone mass, which altogether led the investigators to conclude that there exists a bidirectional signaling between osteoclastic EFNB2 and osteoblastic EphB4.[8] Since the ephrinB ligands and EphB receptors are both membrane proteins, the proposed bidirectional signaling assumes a direct contact between osteoclasts and osteoblasts, which is known to be a rare event on bone sections. In addition, the physiological relevance of osteoclast-derived EFNB2 and osteoblastic EphB4 in vivo remained to be elucidated, since mice with specific deletion of these molecules were not investigated.
Sphingosine-1-phosphate (S1P)
Sphingosine kinase (SPHK) 1 and 2 phosphorylate sphingosine to generate S1P and were identified in osteoclasts, and knockdown of SPHK1 has been shown to slightly enhance osteoclastogenesis.[9] Although osteoclasts themselves express receptors for S1P, S1P1-4, but not S1P5, neither exogenous addition of S1P nor FTY-720, an antagonist of S1P receptors, had any effect on osteoclastogenesis. However, S1P did stimulate the migration of bone marrow macrophages and primary osteoblasts, which express S1P1-3, and the chemotactic effect was inhibited by 2-amino-2-(2-[octylphenyl]ethyl)-1,3-propanediol (FTY-720). Thus, it was proposed that the S1P secreted from osteoclasts has the potential to attract osteoblastic cells, thereby functioning in the communication between osteoclasts and osteoblasts.[9] The findings that SPHK1 expression was induced in mature osteoclasts and that the stimulation by osteoclast conditioned media on mineralization was inhibited by a receptor antagonist 2-amino-N-(3-octylphenyl)-3-(phosphonooxy)-propanamaide (VPC23019) has been confirmed by another group.[10]
Increased expression of SPHK1 was also found in mice with targeted deletion of the cathepsin K gene in osteoclasts, and the conditioned medium from the cethepsin K-deficient osteoclasts contained a modestly increased amount of S1P.[11] The mice lacking cethepsin K in osteoclasts exhibited a higher bone mass with increased bone formation parameters, and the stimulation of ALP activity by the conditioned medium from the cethepsin K-deficient osteoclasts was inhibited by VPC23019, an S1P1,3 receptor antagonist. Thus, it was surmised that the increased secretion of S1P from cathepsin K-deficient osteoclasts is responsible for the stimulation of bone formation in that particular model mouse.[11] However, further studies would seem to be required, such as a thorough bone analysis in a mouse model with specific deletion of the SPHK1 gene in osteoclasts, to elucidate the physiological relevance of osteoclast-derived S1P in bone coupling in vivo.
Platelet-derived growth factor (PDGF)-BB
PDGF, specifically in the form of a BB homodimer, was identified as a factor present in the conditioned medium of osteoclast-like cells generated from a RAW264.7 macrophage cell line with receptor activator of nuclear factor-kappa B ligand (RANKL) that inhibited ALP activity in a mouse osteoblastic cell line (MC3T3-E1) preosteoblastic cell line; the inhibitory activity contained in the conditioned medium was abrogated by a neutralizing anti-PDGF-BB antibody.[12] On the other hand, the conditioned medium of RAW264.7 cells seems to contain a biological activity that attracts MC3T3-E1 preosteoblastic cells, and this chemotactic activity was also detected in the conditioned medium of mature osteoclasts generated from bone marrow macrophages with macrophage colony-stimulating factor (M-CSF) and RANKL.[13] Further, knockdown of PDGF-B gene expression in RAW264.7 cells decreased the chemoattractant activity, which was then rescued by adding recombinant PDGF-BB protein, and in preosteoblastic MC3T3-E1 cells the knockdown of PDGF receptor β, but not PDGF receptor α, reduced the chemoattractant activity derived from RAW264.7 cells.[13] Thus, although these are based on in vitro data and in vivo evidence is lacking, the concept that osteoclast-derived PDGF-BB acts an attractant of preosteoblasts through PDGF receptor β is an interesting hypothesis that warrants further investigation.
Collagen triple helix repeat containing 1 (Cthrc1)
Cthrc1 was originally identified as a gene induced in the aorta 4-8 days after balloon injury; Cthrc1 expression was also induced in NIH3T3 fibroblasts in vitro in response to bone morphogenetic protein (BMP) 4 (BMP-4) or TGF-β, and overexpression of Cthrc1 inhibited type I collagen production and stimulated migratory activity in a smooth muscle cell line.[14] Cthrc1 was then identified as a gene that was induced in an ATDC5 chondrogenic cell line in response to BMP-2; although Cthrc1-null mice did not show any developmental phenotype, the adult mice exhibited a lower bone mass with decreased bone formation, while transgenic overexpression in osteoblasts using a collagen type I promoter resulted in a higher bone mass with elevated bone formation, suggesting that Cthrc1 is a positive regulator of osteoblastic bone formation.[15]
We came across Cthrc1 as a gene that was induced in mature osteoclasts only when they were placed on mineralized matrix, i.e. only when engaged in bone resorption.[7] Recombinant Cthrc1 protein stimulated marrow stromal ST2 cells toward osteoblastic differentiation and inhibited adipogenic differentiation. Although a previous report has shown the expression of Cthrc1 in MC3T3-E1 preosteoblastic cell line, but not in C3H10T1/2 or C2C12 cell line, deletion of the gene in osteoblast lineage cells in vivo using osterix-cre deleter line did not reveal any bone phenotype, whereas osteoclast-specific deletion of the Cthrc1 gene using a cathepsin K-cre knockin deleter mouse resulted in a lower bone mass with reduced bone formation.[7]
These results are consistent with the notion that osteoclast-derived Cthrc1 serves a physiological function in stimulating bone formation and maintaining bone mass. In order to further define the specific function of osteoclast-derived Cthrc1 in the coupling process of bone formation to the preceding resorption in vivo, we adopted a RANKL injection model; following two RANKL injections administered to wild-type mice, osteoclastic bone resorption was rapidly stimulated, as expected, resulting in acute bone loss within 10 days. This catabolic phase was followed by a rather slow anabolic process, with bone mass restored within a period of 8 weeks after the RANKL injections. Importantly, in the mice with the Cthrc1 gene deleted specifically in osteoclasts, the acute phase of osteoclastic bone resorption took place to the same extent as in control mice; however, the subsequent anabolic response was inhibited or delayed, so that at 8 weeks the bone mass was still lower than control mice. We concluded from these results that the Cthrc1 secreted by active osteoclasts functions in the coupling process. To our knowledge, this is the first in vivo demonstration of the physiological function of a particular osteoclast product specifically in the coupling process. Research is in progress in our laboratory to elucidate the mechanism by which the coupling function of Cthrc1 is exerted at the molecular level, including the identity of the receptor complex. It was shown that during mouse development, Cthrc1 was involved in the Wnt/planar cell polarity (PCP) signaling pathway by the formation of a Cthrc1-Wnt-Frizzled (Fzd)/receptor tyrosine kinase-like orphan receptor 2 (Ror2) complex,[16] and since Wnt is a potent anabolic signal in bone,[17] cross-talk between the Cthrc1 and Wnt signals may be of interest. Outside the skeleton, since the expression of Cthrc1 in adult mice is confined to bone and brain, the physiological function of Cthrc1 in cognitive function remains to be determined.
Complement component 3a (C3a)
In searching for humoral factors that are secreted from osteoclasts and stimulate osteoblastic bone formation, we have also taken a biochemical approach using concentrated conditioned medium collected from high-purity osteoclast preparations, and succeeded in identifying complement component C3a by liquid chromatography (LC)-mass spectrometry (MS)/MS analysis.[6] Although the complement system is a systemic defense system and its components are produced in the liver and circulate in the blood, [18] emerging evidence points to a pathophysiological role for local complement activation, such as in osteoarthritis.[19] We detected C3a by enzyme-linked immunosorbent assay (ELISA) in the conditioned medium of osteoclasts; this was unlikely to be due to contamination of serum in the culture medium, since C3a was not detected in the culture medium itself or in the conditioned medium of bone marrow macrophages unless they were induced to differentiate with RANKL. When C3 gene expression was knocked down in osteoclasts, the stimulation of osteoblast differentiation by osteoclast conditioned medium, such as ALP activity, was reduced. C3a acts through binding to a seven transmembrane G protein-coupled receptor, C3aR, which is expressed in osteoblast lineage cells. When C3aR expression in osteoblasts was knocked down, the stimulation of ALP activity by the osteoclast conditioned medium was abrogated, while application of a C3aR agonist stimulated osteoblastic differentiation. Finally, the relevance of C3a-C3aR signaling in bone remodeling was addressed in an ovariectomy (OVX) model. Following OVX, bone resorption is elevated and bone formation is also stimulated to some extent, due to the coupling from resorption to formation, albeit with a net bone loss. When C3aR signaling was suppressed with a receptor antagonist, the elevation of bone formation following OVX was inhibited, with an exacerbation of bone loss.[6] Thus, these results support but do not prove the concept that the C3a derived from osteoclasts is involved in the stimulation of bone formation acting through C3aR. Further studies are required to delineate the specific functions of C3a in osteoclasts and C3aR in the osteoblast lineage by making mouse models with genetic deletion of each molecule in specific target cells.
The coupling of bone formation to resorption is a tightly regulated, complex process involving a multitude of positive and negative regulators. Semaphorin 4D is an example of a negative regulator;[20] these molecules may be positive regulators when osteoclasts disappear from the resorption lacuna, releasing the inhibitory regulation of osteoblasts and allowing bone formation to take place by a newly arrived osteoblast team. Also, a canopy (over the remodeling sites) that is composed of bone lining cells has been proposed as the functional bone remodeling compartment in human bone,[21] but it has rarely been observed in mouse bone, and any factors that emanate from and influence these structures serve as a target for the coupling that occurs between bone resorption and formation. Future research should aim at elucidating the relative importance of these factors and specific physiological and pathological situations in which a certain coupling factors are deployed, such as aging, pregnancy/lactation, immobilization, microgravity and cancer.
 XML Download
XML Download