

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
CATCH22 syndrome is an abbreviation for Cardiac anomaly, Abnormal face, Thymic hypoplasia, Cleft palate, Hypocalcemia, 22q11.2 deletion; it is caused by chromosome 22q11.2 microdeletion and includes DiGeorge syndrome (DGS), conotruncal anomaly face syndrome (CTAFS), and velo-cardio-facial syndrome (VCFS) etc.[1] In Western countries, chromosome 22q11.2 deletion occurs once every 3,000 to 9,000 people. Since clinical phenotypes of DGS, CTAFS, and VCFS are overlapped in many cases, it is difficult to differentiate the syndromes using clinical phenotypes only.[1] It has reported that chromosome 22q11.2 deletion is found in 90% of the patients with DiGeorge phenotype, 70% of the patients with VCFS, and 15% of the patients with isolated conotruncal cardiac defect.[1,2] Most of the patients were diagnosed by slightly and severely decreased immunity, facial defects, and heart anomaly and some patients have kidney abnormalities, hypoparathyroidism, hypothyroidism, and developmental retardation. Therefore, chromosome 22q11.2 deletion is mostly diagnosed in early childhood and childhood and hardly diagnosed in adulthood. So far, 3 cases of adult patients who had CATCH22 syndrome had reported domestically in Korean Journal of Medicine in 1997 and 2006 and in Journal of Korean Endocrine Society in 2012; the patients were 18 and 19 years old in all cases.[3-5] However, CATCH22 syndrome diagnosed in a postmenopausal woman in her 50's without an unusual medical history is very rare enough to be reported only 5 cases all around the world. The objective of this study was to report a case of CATCH22 syndrome with literature review; in a woman in her 50's that visited a hospital due to spasm and was diagnosed by hypocalcemia.
CASE
Patient: 57-year-old female
Chief complaint: Mental change and spasm
Current medical history: Above 57-year-old female patient didn't have an unusual medical history in the past and visited a neurosurgical clinic in another hospital a month before visiting the hospital because she couldn't open both of her hands. With a consideration of Parkinson's disease, she was prescribed with levodopa and then the course was going to be observed in an outpatient clinic. However, the symptom became worse after she started taking levodopa and then she visited the outpatient clinic of the hospital again in a week. The course was going to be examined after stopping to take levodopa but the symptom seemed to get worse gradually even if she stopped taking levodopa. After 5 days, the patient visited an emergency room in the hospital and brain magnetic resonance (MR), brain computed tomography (CT), and C-spine MR were performed; since there was no special opinion with an exception of spinal stenosis, she was discharged from the hospital again. But, the symptom was still lasted; she had been found as tumbling and lying down on the floor a week before visiting the hospital. Since the patient still had the symptoms, she wanted to visit the other hospital. However, she had spasm and a mental change in a car while driving to the other hospital to visit. Then she visited an emergency room in our hospital; she showed low serum calcium, 5.3 mg/dL (reference: 8.2-10.2), in the blood test at that time so she was hospitalized in division of endocrinology and metabolism, department of internal medicine.
Medical history in the past: The patient graduated an elementary school which was not well-educated. From the medical history in the past, no unusual medical history was found in the patient with an exception of taking antihypertensives due to the hypertension that was diagnosed 2 years ago.
Family history: There was no unusual disease or abnormality in the family history.
Physical examination: The patient was in lethargic at the time of visiting the hospital and showed hypocalcemic tetany in both hands with 144 cm of height and 42.6 kg of body weight. There was no noise detected when examining the heart with a stethoscope. Exophthalmos or goiter was not present but she had a high ached plate and a nasal voice. In the neurologic examination, the patient showed positive on Chvostek sign and Trousseau sign and had normal muscle strength.
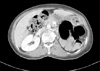
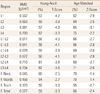
Laboratory test: The tests were performed at the time of visiting our hospital; serum calcium was 5.3 mg/dL (reference value: 8.2-10.2), phosphorus was 5.5 mg/dL (reference value: 2.7-4.5), magnesium was 1.8 mEq/L (reference value: 1.5-2.4), alkaline phosphatase was 89 U/L (reference value: 20-120). Serum parathyroid hormone and 25-OH-Vitamin D3 were 8 pg/mL (reference value: 11-62) and 2.1 ng/mL (reference value: 9.0-37.6), respectively. According to the bone mineral density test utilizing dual energy X-ray absorptiometry (iDXA; GE Healthcare, Madison, WI, USA), indicating that the patient seemed to have osteoporosis (Table 1). Since the patient was considered to have abdominal enlargement, abdominal computerized tomography was carried out and single kidney was observed in the patient (Fig. 1). In the thyroid ultrasonography, no unusual opinion was shown with an exception of thyroid nodule and thyroid hormone seemed to be normal. The patient had a problem with understanding what people said and showed decreased hearing in both ears from the audiometry as the patient appealed. According to Wechsler intelligence test, the intelligence score of the patient was 85 which were below the average and language development was particularly retarded. Since the patient had kidney anomaly, short height, hypocalcemia caused by hypoparathyroidism, decreased hearing, and the retardation of language development, CATCH22 syndrome was considered so that chromosome test was carried out; fluorescence in situ hybridization (FISH) was performed utilizing TUPLE1 probe in the DiGeorge/VCFS region. As a result of the analysis, we were able to confirm diagnosing that the patient had CATCH22 syndrome because the TUPLE1 probe signal that was normally present was not observed in one of the chromosome 22 (Fig. 2). After diagnosing CATCH22 syndrome, Lymphocyte T cell subset was carried out in order to evaluate immunity and the result showed that the patient had normal immunity.
Treatment and course: The patient recovered her consciousness after taking vitamin D and calcium and the low calcium tetany improved. Currently, the patient takes oral vitamin D and calcium (calcitriol 0.5 µg, elemental calcium 800 mg) and the course is still examined. The patient responded well to the treatments and she is on follow-up by outpatient clinic in division of endocrinology and metabolism, department of internal medicine and genetics clinic.
DISCUSSION
Chromosome 22q11.2 deletion syndrome is caused by mircodeletion of chromosome 22q11.2 and inhibition of the development of third and fourth pharyngeal pouches. A decrease and malfunction of T cells can occur; because of that, repetitive infections can occur since early years. The syndrome also shows the symptoms of thymic agenesis, heart anomaly, hypocalcemia caused by hypoparathyroidism , and characterized facial defects etc.[6] It is inheritable but the opposite cases are more common; when parents possess the deletion, their children have 50% chance to possess the deletion.[6,7] In the present case, children and relatives of the patient didn't have any specific genetic disorders. For the pediatric patients with severe heart anomaly and the patients with hypocalcemia in a neonatal period, the syndrome is able to be diagnosed in a neonatal period; on the other hand, if the patients had normal immunity and mild or no heart anomaly and facial defects, the syndrome may not be diagnosed until adulthood. Although the kidney anomaly was observed with single kidney in the present case, CATCH22 syndrome was not diagnosed until the late adulthood of the patient due to the mild facial defect and the normal immunity.
Hypocalcemia caused by hypoparathyroidism is exhibited in 40-60% of patients with chromosome 22q11.2 deletion,[6,7] in 60% of patients with DGS, and 20% of patients with VCFS. In case where hypocalcemia is observed in a neonatal period, the case is strongly considered to be chromosome 22q11.2 deletion. Mostly the syndrome is occurred when a baby is younger than 1 year old, however the cases that adult patients visited hospitals because of hypocalcemia have been reported as well. Especially, most of the patients who were diagnosed chromosome 22q11.2 deletion in adulthood visited hospitals due to spasm caused by hypocalcemia and then were diagnosed the syndrome. The patient in the present case also visited our hospital due to the spasm caused by hypocalcemia; but the case was unique since the first diagnosis was in her 50's which were older than that of in conventional patients.
Kidney deletion related to DGS shows the symptoms of dysmorphic disorders, renal agenesis, polycystic kidney syndrome, stenosis disorders, vesicoureteral reflex, and nephrocalcinosis; medication and surgical treatments are required depending upon the case.[7] The patient in the present case showed the single kidney; the complications caused by the single kidney were not exhibited so that additional treatments were not required.
Typically, 80% of DGS showed a decrease in the number of T cells due to the thymus development disorder; or although the number of cells is normal, decreased immunity such as severe combined immunodeficiency due to malfunction of T cells and insignificant decreased humoral immunity occurs. Besides, repetitive infections occur throughout entire life in 25% of DGS.[8] However, it was reported that most of the patients who had the decreased T cell immunity grew up and possessed normal immunity.[9] The patient in the present case showed normal in the Lymphocyte T cell subset and was estimated to have normal immunity since the medical history had no specific infections in the past.
Since CATCH22 syndrome have relatively high prevalence rate of maximum one out of every 3,000 people and is characterized as autosomal dominant inheritance, genetic counseling in the children is required if diagnosed in the parents. CATCH22 syndrome is mostly diagnosed in early childhood at pediatrics as a congenital disease so that the syndrome is difficult to be considered in the late adulthood like the patient in the present case. However, when facial defects, heart and kidney anomaly, and short height with hypoparathyroidism are present, it is considered to have the tests regarding CATCH22 syndrome.

 XML Download
XML Download