

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Bacterial pathogens are known to possess sophisticated mechanisms for their survival within their hosts by expressing specific products. These products, called virulence factors, are involved in the steps of infectious process, resulting in host-pathogen interaction.12 The outcome of this complex and dynamic interaction is manifested as signs and symptoms of clinical infectious diseases. In cases of root canal and periapical infections, it is widely accepted that anaerobic Gram-negative bacteria are responsible.345 However, the specific roles of putative bacterial pathogens in the pathogenesis of endodontic infections have not yet been adequately assessed. Of these putative pathogens, Enterococcus faecalis (E. faecalis) has been implicated to play an important role in persistent root canal and periapical infections mainly due to its unique presence in these lesions.67
At present, molecular pathogenesis of E. faecalis infection is not well understood, since the virulence factors involved in root canal and periapical infections have not yet been adequately identified or characterized. Several putative virulence factors of E. faecalis with potential of pathogenicity have been described, including aggregation substance, several surface adhesions, sex pheromones, lipoteichoic acid, gelatinase, hyaluronidase, extracellular superoxide production, and bacteriocins.8 However, their exact roles in endodontic infections have not yet been determined since it is unclear whether these virulence factors are involved in the actual disease process. Additional virulence factors from E. faecalis need be identified and characterized to better understand the interactions between this microorganism and the host since they mediate the infectious activities in root canal and periapical infection.
Most traditional bacterial virulence factors have been identified using an in vitro approach in the laboratory. It is well established that these virulence factors may not be involved in the actual disease process since they are not even expressed inside the host.9 It is therefore crucial to identify and/or characterize genes (proteins) expressed in vivo instead of using an in vitro approach, since these in vivo expressed genes (proteins) are more likely to serve as virulence factors. Numerous molecular techniques have been developed for identifying bacterial genes and their products expressed only in the host, including in vivo expression technology, signature-tagged mutagenesis, differential fluorescence induction, transcriptional and proteomic profiling, and in vivo-induced antigen technology (IVIAT).101112131415
Change-mediated antigen technology (CMAT), a modification of IVIAT, is an antibody-based screening technique used to identify bacterial antigens expressed only in vivo when bacteria undergo changes such as an infection.16 This technology uses hyperimmune antisera raised against bacterial cells isolated from infected tissues. The hyperimmune antisera are subsequently adsorbed with cells and extracts of cells that grow in vitro to produce a probe that is reactive only with proteins expressed exclusively in vivo. CMAT has not been used to identify antigens of E. faecalis expressed in vivo. Therefore, the objective of this study was to use CMAT to screen and identify in vivo-expressed antigens of E. faecalis using an experimental animal infection model.
Materials and Methods
Bacterial strains and growth conditions
E. faecalis ATCC 29212 was purchased from American Type Culture Collection (Manassas, VA, USA) and grown on Brain heart infusion (BHI) agar (Becton Dickson, Sparks, MD, USA) at 37℃. For broth growth, bacterial cells were incubated in 10 mL of BHI broth for 12 hours at 37℃. Cell pellets were collected and kept at -80℃ until used. E. coli BL21 (DE3)/pLysS competent cells (Stratagene, La Jolla, CA, USA) were grown overnight on lysogeny broth (LB) agar plates or in LB broth containing kanamycin (30 µg/mL) at 37℃.
Construction of genomic expression libraries of E. faecalis 29212
Genomic DNA from E. faecalis cells were extracted using a G-spin Genomic DNA Extraction Kit for Bacteria (iNtRON Biotechnology, Sungnam, Korea) according to manufacturer's instructions. Purified genomic DNA was then randomly sheared by sonication using a SONOPULS Ultrasonic homogenizer (Bandelin, Berlin, Germany) for 6 seconds at 100% power set of Sonic to generate DNA fragments sized of 1 - 5 kb. After fractionated by agarose gel electrophoresis to remove fragments that were smaller than 1 kb or bigger than 5 kb, DNA fragments of 1 - 5 kb were treated with End-It DNA End-Repair Kit (Epicentre, Madison, WI, USA) and subjected to phenol-chloroform extraction to eliminate any T4 polynucleokinase activity. Expression vector pET-30c (+) (Novagen, Madison, WI, USA) was digested with EcoRV, purified with QIAquick PCR Purification Kit (Qiagen, Valencia, CA, USA), and dephosphorylated with APex heat-labile alkaline phosphatase (Epicentre). A total of 250 ng of pET-30c (+)/EcoRV and blunt-ended E. faecalis DNA fragments at size of 1 - 5 kb were ligated at various molar ratios of vector to insert DNA (1:0.5, 1:1, 1:2, 1:5, and 1:10) using T4 DNA ligase (New England Biolab, Ipswich, MA, USA). The ligated mixture was used to transform E. coli DH5α (Takara, Daejon, Korea). Transformants were selected on LB plates containing kanamycin antibiotics. A total of 10 colonies were randomly selected and screened using colony PCR with T7 promoter primers. The frequency of self-ligation was lower than 20%. Purified recombinant DNA mixture was used to transform E. coli BL21 (DE3)/pLysS Competent Cells (Stratagene). Transformed cells were grown at 37℃ overnight on LB agar plates containing kanamycin (30 µg/mL).
Experimental endodontic infection
Animal experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee, Chonnam National University (approval number: YB-2011-19). All surgical procedures were performed under sterile conditions. One 3 year old male beagle dog with weight of 10 kg was used for this study. Anesthetic induction was achieved by intravenous administration of thiopental (13.2 mg/kg body weight) followed by administration of 1 - 2% of isoflurane via an endotrachial tube. Additional local anesthesia of 2% lidocaine hydrochloride with 1:100,000 epinephrine was used to give a regional block. Preoperative radiograph was taken to determine any existing periodontal or endodontic lesion. After the induction, the second, third, and fourth premolars of each side of the upper jaw were used for inducing experimental endodontic infection as follows. A high-speed handpiece equipped with a round bur with saline irrigation was used to perforate a buccal surface of each tooth until it reached the pulp tissue. A total of 1.5 × 105 E. faecalis cells were then injected into the pulp cavity using an insulin syringe to induce endodontic infection. The access opening on the buccal surface was subsequently covered with light-cured composite materials. After the surgical procedure, analgesic medication (3 mg/kg body weight, Ketopro, Unibiotech Co., Yesan, Korea) was administered for three days.
Three weeks after injecting bacterial cells into the pulp, a radiograph was taken again to assess the status of periapical lesion of infected teeth. At this time, the dog was anesthetized again using the same protocol described above. All infected premolars were extracted and hemisectioned. Infected pulp and root canal tissues were removed using a barbed broach. They were immediately dissolved in phosphate buffered saline (PBS). Bacterial cell pellets were obtained by centrifugation at ×4,000 g for 20 minutes as described previously.17 Purified cell pellets were immediately stored in -20℃. They were sent to a commercial vendor (Young In Frontier Co., Seoul, Korea) to produce hyperimmune antisera in rabbits. Based on optical density, it was determined that the bacterial cell pellets contained more than 5 × 109 cells. The presence of E. faecalis was confirmed by visual inspection under a light microscope and sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis. The efficacy of produced antibodies was monitored by the enzyme-linked immunosorbent assay (ELISA) analysis.
Serum adsorption
To obtain a serum probe that reacted only to in vivo expressed E. faecalis antigens, hyperimmune rabbit sera were adsorbed with E. faecalis cells cultivated in vitro. Adsorptions were performed using whole bacterial cells. First, rabbit polyclonal antiserum was mixed with 1 × 109 E. faecalis cells with slow agitation (30 rpm) at 4℃ for 1 hour. Cells were removed by centrifugation at ×5,000 g for 10 minutes and the serum (supernatant) was recovered. This step was repeated 4 times. After centrifugation, adsorbed serum was retrieved and stored at -80℃ until used. Cell lysates bound to nitrocellulose membrane were added into serum after removing whole cells and agitated overnight at 4℃. The serum was collected the next day. To remove antibodies that bound to E. coli proteins, the serum was agitated again with nitrocellulose membrane-bound E. coli BL21 harboring pET-30c(+) vector only.
To determine the adsorption efficiency, ELISA was performed as described previously.18 Briefly, a 96-well plate was coated with E. faecalis crude protein extracts at a concentration of 0.1 mg/100 mL in a coating buffer (0.1 M sodium carbonate, pH 9.6) overnight at 4℃. After washing, a blocking solution (2% skim milk in PBS, 300 µL) was added and incubated at 37℃ for 1 hour, followed by washing with PBS. The adsorbed rabbit antisera was added to each well using a serial dilution starting from 1:1,000 dilution and incubated at 37℃ for 2 hours. After the incubation, the plate was washed three times with 1X Tris buffered saline and Tween (TBS-Tween). Then, 50 mL of 1:10,000 diluted goat anti-human IgG conjugated with horse radish peroxidase (Abcam, Hanam, Korea) was added to each well and incubated at 37℃ for 1 hour. After washing the wells three times with TBS-Tween solution, TMB solution (GenDEPOT, Barker, TX, USA) was added to each well. After incubating at room temperature for 2 minutes, the reaction was stopped by adding 1 N H2SO4 (100 mL). Absorbance was measured at 450 nm using a 96-well plate reader (spectrometer).
Genomic library screening for E. faecalis proteins uniquely expressed in vivo
E. coli recombinant clones were screened using the adsorbed hyperimmune rabbit sera as described previously.19 Briefly, kanamycin-resistant E. coli clones (approximately 200 per plate) were transferred to a nitrocellulose membrane. In vivo grown E. faecalis and E. coli containing pET-30c (+) were spotted onto the membrane as positive and negative control for screening, respectively. The membrane was then transferred (colony side up) to a LB agar plate containing 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) to induce protein expression. After incubation at 37℃ for 3 hours, the membrane was removed and the attached cells were lysed by exposure to chloroform vapor. The membrane was then blocked with 5% skim milk in PBS for 1 hour, washed with PBS-Tween 20, and incubated overnight with the adsorbed rabbit hyperimmune serum (1:500) at 4℃. After the incubation, the membrane was washed and probed with goat anti-rabbit IgG conjugated with horse radish peroxidase (1:1,000) at room temperature for 1 hour. A 1-step chloronaphthol (4CN, Thermo Scientifc, Waltham, MA, USA) was used as substrate to identify reactive clones. Positive clones showing reactivity were isolated from the original (master) plate. They were grown on LB-kanamycin plates. Their reactivity was confirmed again using a 4CN as substrate.
Identification of genes isolated by CMAT
Plasmid DNAs from positive clones were purified using a QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA, USA). DNA sequences of inserted DNA fragments from E. faecalis were determined by direct sequencing using a T7 promoter primer with an ABI Prism 377 automatic DNA sequencer by double-strand dideoxy chain termination method at GenoTech Corp. (Daejeon, Korea). Identified gene sequences were compared to DNA and protein databases using a BLAST program (http://www.ncbi.nlm.nih.gov) and analyzed by Vector NTI Software (Invitrogen, Carlsbad, CA, USA). Functional classification of identified antigens was based on published studies of identified proteins of E. faecalis if available.
Results
Adsorption of rabbit hyperimmune antisera
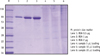
SDS-PAGE analysis confirmed the presence of bacterial cells in the sample acquired from experimental endodontic infection (Figure 1). The hyperimmune rabbit antisera produced against E. faecalis infected pulp and periapical tissues were successively adsorbed against E. faecalis cells grown in vitro. The adsorption efficiency was determined by examining immuno-reactivity of antisera using ELISA after each adsorption step with in vitro grown E. faecalis. Fully adsorbed serum exhibited a significant lower reactivity to in vitro grown E. faecalis (Figure 2), suggesting that the adsorption process efficiently removed antibodies against most E. faecalis proteins produced during in vitro cultivation.
Genomic library construction and screening
Plasmid DNAs of ten colonies randomly selected from the library were subjected to direct DNA sequencing to confirm the presence of the DNA inserts present in the recombinant plasmids. Of the 10 colonies, eight had DNA inserts in the pET-30c (+) vector. A total of approximately 4,500 recombinant clones were screened using the adsorbed pooled serum. In vivo grown E. faecalis cells and E. coli cells containing only pET-30c (+) placed on the nitrocellulose membrane was used as positive and negative control, respectively, for the immunoscreening. Initial screening resulted in the detection of 30 immuno-reactive clones. Of the 30 clones, nineteen reproducibly exhibited reactivity with the adsorbed serum. Therefore, they were subjected to further DNA sequencing.
Identification and functional classification of E. faecalis genes identified by CMAT
Sequencing of the DNA inserts of the 19 positive clones revealed 16 open reading frames (ORFs). Sequences of the remaining three clones could not be obtained. Sequences of the 16 clones were analyzed using BLAST program. CMAT-identified in vivo-induced genes and their predicted functions are listed in Table 1. Many determined gene sequences contained portions of ORFs for putative genes. The size of ORF varied from 663 bp to 2,911 bp. In addition, multiple ORFs were not identified. Sequence analysis revealed that many of these ORFs were enzymes implicated in housekeeping functions with intermediary metabolism. In addition, conserved hypothetical proteins with unknown function and putative membrane protein were also identified, along with copper resistance proteins.
Discussion
Very limited information regarding the nature of virulence factors of E. faecalis has hampered our understanding of the mechanisms by which this endodontic pathogen interacts with the host and ultimately leads to endodontic infection. Considering that genes (proteins) expressed in vivo are very likely to be important in the microorganisms' ability to survive in the host and/or serve as virulence factors, our major objective was to identify E. faecalis proteins that were uniquely expressed in vivo during the actual infectious process. For this purpose, we adopted a new technique called CMAT designed to detect immunoreactive proteins exhibiting specificity only to in vivo-induced proteins. For this immunoscreening, we needed to prepare a 'probe' antibody that reacts only to in vivo-induced proteins (not proteins produced in vitro). CMAT is a modification of the IVIAT that has enabled many investigators to identify in vivo-induced proteins directly from the human host rather than from a potentially misleading animal model.1519 IVIAT has been successfully used to study pathogens such as Candida albicans, Vibrio cholera, Mycobacterium tuberculosis, Vibrio vulnificus, Escherichia coli, Salmonella enterica, Group A Streptococcus, and periodontal pathogens including Aggregatibacter actinomycetemcomitan, Porphyromonas gingivalis, and Tannerella forsythia.18202122232425262728 However, the use of IVIAT requires high antibody titers in human sera against the etiological agents, it may not be readily applicable to the study of microorganisms that do not induce strong humoral responses in humans. Therefore, CMAT was specifically developed to address the above limitation of IVIAT.
The original design of this study was to use E. faecalis samples from clinical cases of pulp and endodontic infection from human subjects. However, we failed to secure adequate amount of E. faecalis cells from clinical cases that could induce sufficient humoral immune responses (data not shown). Therefore, as an alternative, we adopted an animal model using a beagle dog for the purpose of producing in vivo-grown E. faecalis cells. In this model, pulp tissues were intentionally infected with E. faecalis cells in hopes of causing endodontic infection. From the lesions of pulp and root canal infections, we were able to collect adequate amount of E. faecalis cells for the CMAT purpose.
In this pilot study, we used CMAT to identify more than a dozen protein antigens that were reactive with adsorbed antisera reactive with in vivo-grown E. faecalis cells. To the best of our knowledge, this is the first attempt to identify in vivo-induced proteins from E. faecalis. One of the advantages of using CMAT is that it does not require an efficient genetic manipulation system nor human infection because it can utilize an alternative animal model.
Many in vivo-induced genes of E. faecalis identified by CMAT included several enzymes related to housekeeping functions, including energy metabolism, translation, amino acid biosynthesis, and other cellular process (Table 1). These genes were similar to those identified by previous IVIAT studies.1829 These genes might play roles that are not required for in vitro growth.30 It has been observed that 'housekeeping' genes are not constitutively expressed at the transcriptional level in bacteria as previously assumed, indicating that housekeeping metabolism as a very dynamic process is extremely capable of adapting to different growth conditions.313233 Therefore, it is plausible that these genes are up-regulated and expressed only in vivo.
A copper resistance protein was identified in this study. Although its exact function is unknown, it has been found in other bacteria including E. coli, Pseudomonas syringae, and Xanthomonas axonopodis.34 Considering metallic copper can act as antimicrobial surface, copper resistance could function as a potential virulence factor.35 CMAT was able to identify several putative membrane proteins. Since little is known regarding the functions of membrane protein in E. faecalis, analysis of these in vivo induced proteins identified in this study may increase our understanding on the pathogenic mechanisms of this microorganism.
Further use of CMAT in E. faecalis will be required to identify additional in vivo induced proteins that are likely to be important in survival in the host and may serve as virulence factors in the disease process. In future studies, the functional roles of these identified genes can be tested by constructing isogenic mutants for use in animal models or in vitro models. These efforts will ultimately allow us to gain a better insight into the pathogenic mechanisms of E. faecalis. For example, gene expression (mRNA) analysis using real-time PCR for each identified gene could be performed to confirm its in vivo expression.1836 Genetic analysis using isogenic mutant (such as siRNA-knockdown or gene destruction) could be performed to confirm the identified genes (proteins) are really related to the disease process, as previously performed.3738
Once functional roles are confirmed to be important for E. faecalis virulence, these virulence factors could be analyzed as candidates for diagnostic, therapeutic, and preventive measures for endodontic infections. Identified virulence factors could be used as disease markers for pulpal and peri-apical infection associated with E. faecalis. The presence of these virulence factors in the pulp and periaplical tissues will confirm the infectious disease process mediated by E. faecalis without bacterial culture of the clinical samples. Another potential application includes the development of therapeutic and preventive approaches targeting these virulence factors, such as intracanal medication, vaccines, and novel antimicrobial substances.22 By specifically inactivating virulence factors without affecting other non-virulent components, it would be possible to effectively inhibit or reduce virulence mediated by E. faecalis. These efforts will lead to improved management of persistent endodontic infections associated with E. faecalis.
Conclusions
Sixteen in vivo-induced proteins of E. faecalis were identified using CMAT and experimental endodontic infection model. These proteins were expressed uniquely in vivo and reactive with sera produced against in vivo-grown E. faecalis. The results of this pilot study suggest that CMAT adopting an experimental animal model of endodontic infection is a useful approach to identify and characterize potential virulence factors of E. faecalis. Detailed analysis of these in vivo-induced genes (proteins) will lead to a better understanding of the molecular mechanisms involved in the endodontic infection of E. faecalis.


 XML Download
XML Download