

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Magnesium is the major intracellular divalent cation and plays an essential physiological role in many functions of the body1). Magnesium is essential for the synthesis of nucleic acids and proteins, and is an important cofactor for a wide range of enzymes, transporters. Magnesium has important effects on the cardiovascular system. Intracellular magnesium forms a key complex with ATP and has a key role in many other important biological processes such as protein synthesis, cell replication, and energy metabolism2).
Go to : 
Magnesium metabolism
1. Body content and distribution of magnesium
The normal adult human body contains approximately 1,000 mmols of magnesium (22-26 g). The distribution of magnesium within the body is shown in Fig. 13).
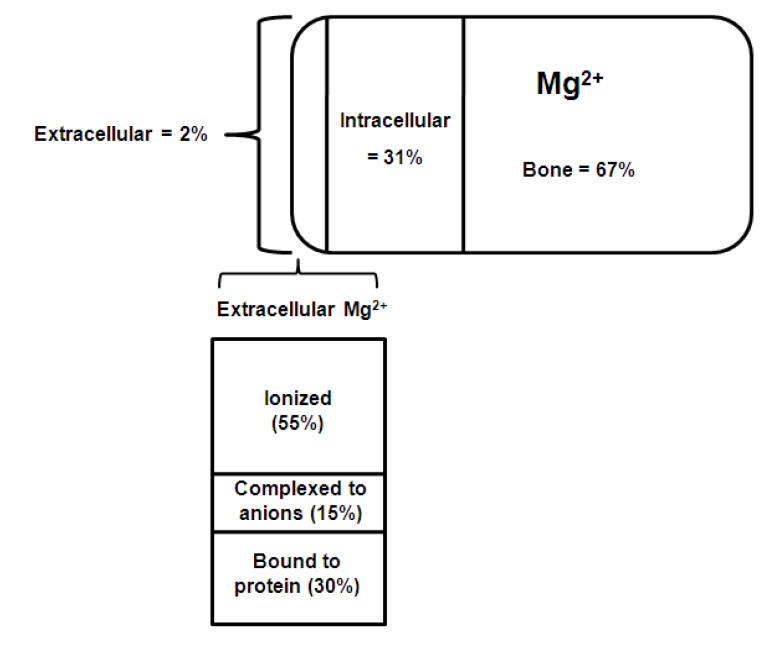 | Fig. 1Distribution of chemical forms of magnesium in serum. Of total body magnesium, 67% is found in bone and hard tissue; 31% is found inside cells; and approximately 2% is found in serum3).
|
About 60% of the magnesium is present in bone, of which 30% is exchangeable and functions as a reservoir to stabilize the serum concentration. About 20% is found in skeletal muscle, 19% in other soft tissues and less than 1% in the extracellular fluid. Intracellular magnesium is maintained within narrow concentration limits except in extreme situations such as hypoxia or prolonged magnesium depletion. Very little is known about the mechanisms involved in the regulation of intracellular magnesium4).
Go to :
Magnesium balance
The recommended daily allowance (RDA) for magnesium in adults is 4.5 mg/kg/day, lower than the previous recommendation of 6-10 mg/kg/day. The daily requirement is higher in pregnancy, lactation and following debilitating illness. Recent dietary surveys show that the average intake in many western countries is less than the RDA5). Magnesium intake depends on the magnesium concentration in drinking water and food composition. Magnesium is plentiful in green leafy vegetables such as spinach and broccoli (which are rich in magnesium-containing chlorophyll), cereal, grain, nuts, banana, and legumes. Fruits, meats, chocolates, and fish have intermediate values, and dairy products are poor in magnesium5, 6). Fig. 2 illustrates the metabolism of magnesium in a healthy adult1). The average magnesium intake of a normal adult is approximately 12 mmol/day. In addition to this, approximately 2 mmol/day of magnesium is secreted into the intestinal tract in bile and pancreatic and intestinal juices. From this pool 6 mmol (about 30%) is absorbed giving a net absorption of 4 mmol/day.
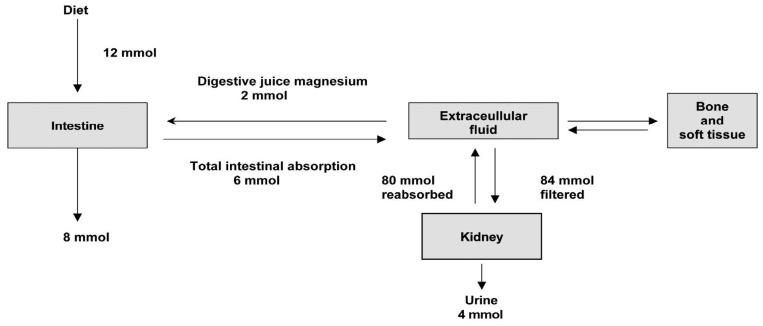 | Fig. 2Distribution of magnesium in the body1).
|
Go to :
Intestinal absorption of magnesium
About 30-40% of the dietary magnesium content is absorbed, mainly in the jejunum and ileum. Fractional intestinal absorption of magnesium is inversely related to intake; 65% at low intake and 11% at high intake. Most of the absorption occurs in the jejunum, ileum, and colon. At normal intake, absorption is primarily passive. During low magnesium intake a saturable component of magnesium absorption can be demonstrated7). Factors controlling magnesium absorption are not well understood. Studies suggest a role for parathyroid hormone (PTH) in regulating magnesium absorption. Intestinal magnesium absorptive efficiency is stimulated by 1,25-dihydroxyvitamin D (1,25(OH)2D) and can reach 70% during magnesium deprivation, but the role of vitamin D and its active metabolite 1,25(OH)2D is controversial1).
Go to :
Magnesium reabsorption in the kidney
The kidney plays a major role in magnesium homeostasis and the maintenance of plasma magnesium concentration. Urinary magnesium excretion normally matches net intestinal absorption and is ~4 mmol/d (100 mg/day). Regulation of serum magnesium concentration is achieved mainly by control of renal magnesium reabsorption. Under normal circumstances, when 80% of the total plasma magnesium is ultrafiltrable, 84 mmol of magnesium is filtered daily and 95% of this reabsorbed, leaving about 3-5 mmol to appear in the urine. Under normal circumstances, approximately only 20% of filtered magnesium is reabsorbed in the proximal tubule, whereas 60% is reclaimed in the cortical thick ascending limb (TAL) of loop of Henle and the remaining 5-10% in the distal convoluted tubule (DCT)8, 9).
Magnesium transport in the proximal tubule appears to be primarily a uni-directional passive process depending on sodium/water reabsorption and the luminal magnesium concentration. Magnesium transport in the TAL is directly related to sodium chloride reabsorption and the positive luminal voltage in the segment4). In the TAL, approximately 25% of the filtered sodium chloride is reabsorbed through the active transcellular transport (sodium-chloride-potassium transport) and passive paracellular diffusion10). This creates a favorable luminal positive potential at the TAL where most of the magnesium is reabsorbed. Magnesium reabsorption in the TAL occurs via a paracellular route that requires both a lumen-positive potential, created by NaCl reabsorption, and the tight-junction protein, claudin-16/paracellin-1.
The mutations in claudin-16/paracellin-1 are responsible for familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC). Magnesium reabsorption in the TAL is increased by PTH but inhibited by hypercalcemia, both of which activate the calcium sensing receptor (CaSR) in this nephron segment. Magnesium reabsorption in the DCT is active and transcellular11, 12).
The active transcellular transport of magnesium in the DCT was similarly enhanced by the realization that defects in transient receptor potential melastatin 6 (TRPM6) cause hypomagnesemia with secondary hypocalcemia (HSH). This channel regulates the apical entry of magnesium into epithelia and alters whole-body magnesium homeostasis by controlling urinary excretion. TRPM6 is regulated at the transcriptional level by acid-base status, 17β-estradiol, and both FK506 and cyclosporine. The molecular identity of the protein responsible for the basolateral exit of magnesium from the epithelial cell remains unidentified13). Factors influencing renal magnesium excretion are listed in Table 2. The plasma magnesium concentration is a major determinant of urinary magnesium excretion. Hypomagnesemia is associated with an increase in magnesium excretion due to an increase in the filtered load and reduced reabsorption in TAL1).
Go to :
Assessment of magnesium status
At present, there is no simple, rapid, and accurate laboratory test to indicate the total body magnesium status. The most commonly used method for assessing magnesium status is the serum magnesium concentration. The total serum magnesium concentration is not the best method to evaluate magnesium status as changes in serum protein concentrations may affect the total concentration without necessarily affecting the ionized fraction or total body magnesium status. The correlation between serum total magnesium and total body magnesium status is poor5, 14).
Measurement of ultrafiltrable magnesium may be more meaningful than the total magnesium as it is likely to reflect ionized magnesium concentration, but methods are not available for routine use. In the last few years, ion selective electrodes for magnesium have been developed and several commercial analyzers are now available for the measurement of ionized magnesium concentration.
Measurement of ionized magnesium has been found to be useful in several clinical situations. In summary, no single method is satisfactory to assess magnesium status. The simplest, most useful, and readily available tests are the measurement of serum total magnesium and the magnesium tolerance test3, 5). Ionized magnesium measurement may become more readily available with the development of reliable analyzers.
Go to :
Hypomagnesemia and magnesium deficiency
The terms hypomagnesemia and magnesium deficiency are commonly used interchangeably. However, total body magnesium depletion can be present with normal serum magnesium concentrations and there can be significant hypomagnesemia without total body deficit1). Hypomagnesemia is frequently undetected. Measurement of serum magnesium concentration in 1,000 samples received for electrolyte determination showed that only 10% of the hypomagnesaemic patients had magnesium requested15). Thus, it has been suggested that magnesium should be determined routinely in all acutely ill patients especially in those with conditions, diseases, or treatment that may predispose to magnesium deficiency11).
1. Etiology and pathogenesis of hypomagnesemia
Hypomagnesemia may result from one or more of the following mechanisms: redistribution, reduced intake, reduced intestinal absorption, increased gastrointestinal loss, and increased renal loss.
1) Hypomagnesemia due to redistribution
Hypomagnesemia due to the shift of magnesium from extracellular fluid into cells or bone is seen in refeeding of starved patients (refeeding syndrome), during treatment of metabolic acidosis, and in hungry bone syndrome which is seen after parathyroidectomy or in patients with diffuse osteoblastic metastases1).
2) Gastrointestinal causes
Magnesium deficiency entirely due to reduced dietary intake in otherwise healthy subjects is very uncommon. Hypomagnesemia may be seen in patients who are maintained on magnesium-free intravenous fluids or total parenteral nutrition, especially in those patients who have a marginal or reduced serum magnesium to start off with11). An inherited disorder of isolated magnesium malabsorption associated with hypocalcemia, tetany, and seizures has been described in infants as well as in older individuals16). Children with this condition usually present at 4-5 weeks of age with generalized convulsions associated with protein losing enteropathy, hypoalbuminaemia, and anasarca. The disorder is caused by a mutation in the TRPM6 gene which codes for an ion channel, resulting in defective carrier mediated transport in the small intestine10).
3) Renal causes
Proximal tubular magnesium reabsorption is proportional to sodium reabsorption, and a reduction in sodium reabsorption during long-term intravenous fluid therapy may result in magnesium deficiency1).
(2) Inherited disorders
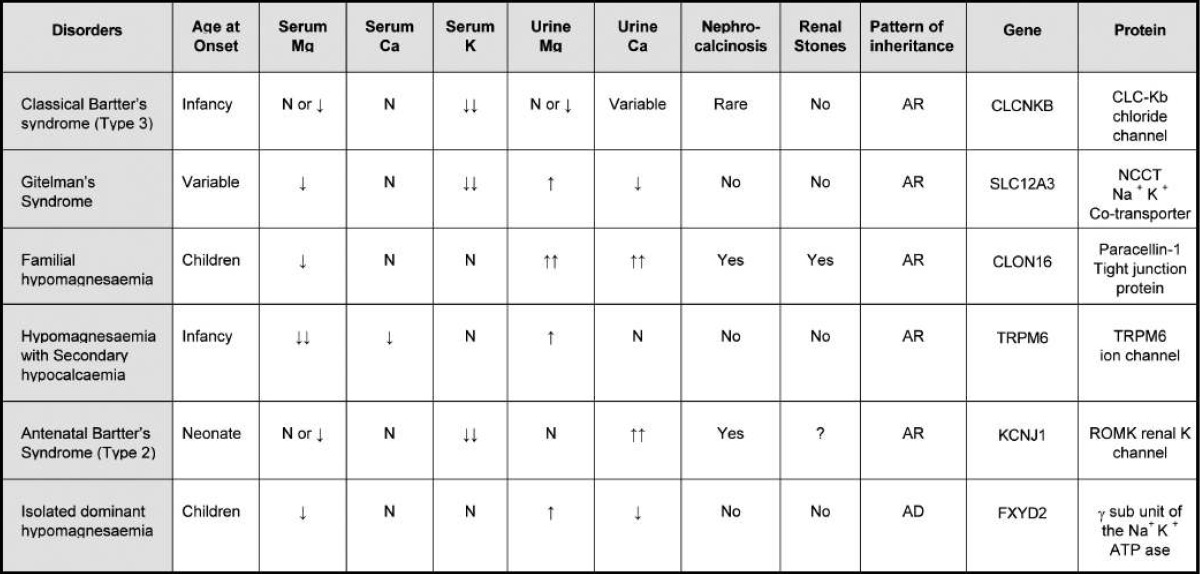
Several different congenital disorders of renal tubular reabsorption of magnesium have been described but there is no consensus on their classification9, 10). The classification, features and molecular defects in some of the syndromes are listed in Table 21). And renal regulation of magnesium homeostasis is illustrated in Fig. 313).
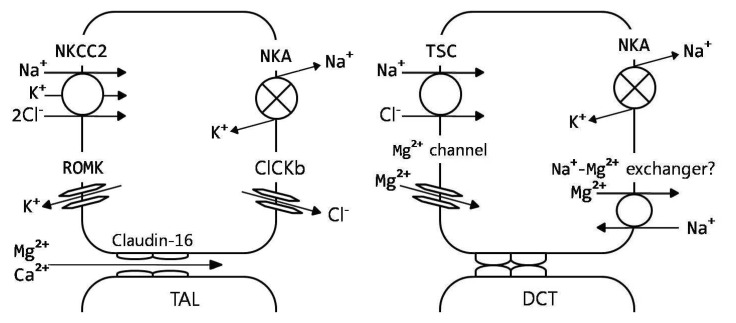 | Fig. 3Renal regulation of magnesium homeostasis. Model of thick ascending limb (TAL) and distal convoluted tubule (DCT), showing predominant magnesium transport pathways9). CLCKb, voltage-dependent chloride channel; claudin-16, paracellin; NKA, Na+,K+-ATPase; NKCC2, Na+/K+/2Cl- cotransporter; ROMK, inwardly rectifying potassium channel; TSC, thiazide-sensitive Na+/Cl- cotransporter.
|
4) Drugs
A variety of drugs including antibiotics and chemotherapeutic agents cause magnesium wasting. Loop diuretics inhibit magnesium transport in TAL and cause magnesium depletion, especially during long-term use19, 20). Short-term administration of thiazide diuretics which act on the DCT, where less that 5% of magnesium is absorbed, does not produce magnesium wasting. However, long term administration may produce substantial magnesium depletion due to secondary hyperaldosteronism, increased sodium load, and interaction with calcium metabolism21).
Hypomagnesemia is a frequent complication of cisplatin treatment and may be acute or chronic. Cisplatin, for example, frequently causes magnesium wasting with hypocalciuria and hypokalemia, resembling Gitelman syndrome22), a disease caused by defective electroneutral NaCl cotransporter in the DCT. Cisplatin increases the transepithelial voltage in the DCT23), consistent with a 'Gitelman-like' effect. The incidence of hypomagnesemia increases with cumulative dose. In the acute phase, poor dietary intake and the use of diuretics are contributing factors. Chronic hypomagnesemia starts to develop 3 weeks after the initial chemotherapy and usually persists for several months24).
Go to :
Manifestations of hypomagnesemia and magnesium deficiency
Many patients with hypomagnesemia and magnesium deficiency remain asymptomatic. As magnesium deficiency is usually secondary to other disease processes or drugs, the features of the primary disease process may complicate or mask magnesium deficiency. Signs and symptoms of magnesium deficiency are usually not seen until serum magnesium decreases to 0.5 mmol/L or lower1). Manifestations may depend more on the rate of development of magnesium deficiency and/or on the total body deficit rather than the actual serum magnesium concentration18). Clinical manifestations of severe or moderate magnesium deficiency are listed in Table 3.

Go to :
Management of hypomagnesemia and magnesium deficiency
1. Prophylaxis of hypomagnesemia
In patients likely to develop magnesium deficiency, prophylactic measures should be taken to prevent the development or progression of hypomagnesemia and magnesium deficiency. High-risk patients such as chronic alcoholics, patients receiving total parenteral nutrition, long term diuretic therapy or other drugs causing magnesium loss, and those with chronic diarrheal and steatorrheal states should have serum magnesium monitored regularly and, if necessary, supplemented with magnesium. Patients on parenteral nutrition should receive prophylactic doses of 4-8 mmol/day of magnesium. Higher doses may be required in malnourished patients and in those with ongoing magnesium loss1).
2. Treatment of hypomagnesemia
Mild, asymptomatic hypomagnesemia may be treated with oral magnesium salts [MgCl2, MgO, Mg(OH)2] in divided doses totaling 20-30 mmol/d (40-60 mEq/d). Diarrhea may occur with larger doses12). Assessment of renal function before replacement therapy is important, and magnesium therapy in patients with any renal failure should be undertaken cautiously. During intravenous replacement therapy it is important to monitor serum concentrations of magnesium, potassium, and other major cations as well as deep tendon reflexes. If there is deterioration in renal function, the dose of magnesium should be halved and serum magnesium monitored more frequently.
If hypermagnesemia or signs of magnesium intoxication (hypotension, bradycardia or depression of tendon reflexes) develop, therapy should be stopped1). More severe hypomagnesemia should be treated parenterally, preferably with MgCl2, which can be administered safely through a continuous infusion of 50 mmol/d (100 mEq Mg2+/d) if renal function is normal. MgSO4 may be given IV instead of MgCl2, although the sulfate anions may bind calcium in serum and urine and aggravate hypocalcemia. Serum magnesium should be monitored at intervals of 12-24 hours during therapy, which may continue for several days because of impaired renal conservation of magnesium (only 50-70% of the daily IV magnesium dose is retained) and delayed repletion of intracellular deficits, which may be as high as 1-1.5 mmol/kg (2-3 mEq/kg)12).
Go to :
Hypermagnesemia
Hyermagnesemia is rarely seen in the absence of renal insufficiency, as normal kidneys can excrete large amounts (250 mmol/d) of magnesium. Prevalence of Hypermagnesemia varies from 5.7% to 9.3%15, 25). The highest serum magnesium concentrations reported so far are 18 mmol/L in a 33 week old premature infant and 13.4 mmol/L in a 78 year old woman who swallowed water from the Dead Sea25, 26). Severe hypermagnesemia in fact seems to be a feature in patients who drown in the Dead Sea26).
1. Causes of hypermagnesemia
A notable example of a cause of hypermagnesemia is prolonged retention of even normal amounts of magnesium-containing cathartics in patients with intestinal ileus, obstruction, or perforation. Extensive soft tissue injury or necrosis can also deliver large amounts of magnesium into the ECF in patients who suffered trauma, shock, sepsis, cardiac arrest, or severe burns12). Hypermagnesemia commonly occurs due to the excessive administration of magnesium salts or magnesium-containing drugs, especially in patients with reduced renal function. Hypermagnesemia may rarely be due to redistribution from cells27). (Table 4)

1) Excessive intake
Magnesium containing medications are commonly used as laxatives, antacids, and as rectal enemas. Hypermagnesemia has often been described with the use of magnesium containing cathartics for treatment of drug overdose, in patients taking magnesium-containing cathartics and antacids for therapeutic purposes and following rectal administration of magnesium, even in the presence of normal renal function28).
2) Renal failure
Hypermagnesemia is common in patients with end stage renal disease, in those undergoing dialysis and in acute renal failure27). In chronic renal failure, serum magnesium concentration is usually maintained until the glomerular filtration rate falls below 30 mL/min. However, severe hypermagnesemia may result, especially if magnesium-containing medications are used. In patients undergoing regular dialysis, the serum magnesium concentration is directly related to the dialysate magnesium concentration. Hypermagnesemia is also seen in patients undergoing continuous ambulatory peritoneal dialysis (CAPD)29).
Go to :
Effects of hypermagnesemia
Signs and symptoms of hypomagnesemia are not usually apparent until serum magnesium is in excess of 2 mmol/L; however, the serum concentration at which signs and symptoms appear varies widely1). (Table 5)

1. Neuromuscular manifestations
The most prominent clinical manifestations of hypermagnesemia are vasodilation and neuromuscular blockade, which may appear at a serum magnesium concentration of greater than 2 mmol/L (>4 mEq/L; >4.8 mg/dL)12). Neuromuscular symptoms are the most common presenting problem of hypermagnesemia and magnesium intoxication. Magnesium prevents the release of pre-synaptic acetylcholine from both sympathetic and neuromuscular junctions26). Hypermagnesemia causes blockage of neuromuscular transmission and depresses the conduction system of the heart and the sympathetic ganglia. Clinically, one of the earliest effects of magnesium intoxication is the disappearance of deep tendon reflexes. This is often seen at magnesium concentrations of 2.0-4.5 mmol/L. Somnolence may be observed at serum concentrations of 2 mmol/L or above1).
2. Cardiovascular manifestations
Hypotension, refractory to vasopressors or volume expansion, may be an early sign of hypermagnesemia12). A moderate increase in serum magnesium concentration of 2-3 mmol/L results in mild reduction in supine as well as erect blood pressure. A higher concentration of magnesium may cause severe symptomatic hypotension30). Paradoxical bradycardia; prolongation of PR, QRS, and QT intervals; heart block; may be seen at serum magnesium levels approaching 10 mmol/L, asystole12).
3. Hypocalcaemia
Magnesium intoxication causes a reduction in serum calcium concentration. This has most commonly been reported in patients receiving magnesium therapy for pregnancy-induced hypertension31). Hypermagnesemia, acting via the CaSR, causes hypocalcemia and hypercalciuria due to both parathyroid suppression and impaired cortical TAL calcium reabsorption12).
4. Other effects
Hypermagnesemia may cause smooth muscle paralysis resulting in paralytic ileus. Other non-specific manifestations of magnesium intoxication include nausea, vomiting, lethargy, and weakness may progress to respiratory failure and paralysis. Other findings may include gastrointestinal hypomotility or ileus; facial flushing and papillary dilation12).
Go to :
Management of hypermagnesemia
Most cases of hypermagnesemia can be prevented. The possibility of hypermagnesemia should be anticipated in any patient receiving magnesium treatment, especially if the patient has reduced renal function, serum magnesium concentration should be monitored daily. When hypermagnesemia is found, we should identify and interrupting the source of magnesium and magnesium therapy should be withdrawn. This is all that is needed in most patients with mild to moderate hypermagnesemia1).
Use of magnesium-free cathartics or enemas may be helpful in clearing ingested magnesium from the gastrointestinal tract. Vigorous IV hydration should be attempted, if appropriate. Hemodialysis is effective and may be required in patients with significant renal insufficiency. In patients with symptomatic hypermagnesemia, serum magnesium should be lowered and the effects of hypermagnesemia antagonized. Calcium, administered IV in doses of 100-200 mg over 1-2 hours, has been reported to provide temporary improvement in signs and symptoms of hypermagnesemia12).
Calcium antagonizes the toxic effects of magnesium and therefore patients with severe magnesium intoxication should be given 1 gm of intravenous calcium gluconate. This should be followed by the infusion of 150-100 mg of calcium over 5-10 minutes, which usually causes a dramatic improvement in the patient's clinical condition. Administration of glucose and insulin may also help to promote magnesium entry into cells. If the patient is in renal failure, peritoneal or hemodialysis against a dialysate with low magnesium levels will rapidly and effectively lower the serum magnesium concentration1).
Go to :
Summary
Disorders of the magnesium metabolism are relatively common in hospital patients. Magnesium balance in the body is controlled by a dynamic interplay among intestinal absorption, exchange with bone, and renal excretion. Intestinal magnesium absorption proceeds in both a passive paracellular and an active transcellular manner. Regulation of serum magnesium concentrations is achieved mainly by control of renal magnesium reabsorption. Only 20% of filtered magnesium is reabsorbed in the PT, whereas 60% is reclaimed in the cortical TAL and another 5-10% in the DCT. The passive paracellular transport of magnesium in the TAL is closely related with claudin-16/paracellin-1. The active transcellular transport of magnesium in the DCT is similarly enhanced by the active transcellular transporter, TRPM6.
Go to :

 XML Download
XML Download