

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Inorganic phosphate (Pi) is essential for various cellular metabolism and skeletal mineralization. It is an essential part of nucleic acids and the cell membrane, serves as an important mediator of intracellular signaling, and regulates protein activity. About 600 g (500-700 g) of phosphorus is present in normal adults, of which 80% to 85% is present in bone mineral. In serum, most of the phosphorus is present as Pi in normal concentration of 0.75 to 1.45 mmol/L (2.5 to 4.5 mg/dL). More than 85% of Pi in serum is present as the free ion and less than 15% is protein-bound. Free HPO42- and NaHPO4- predominantly account for ~75% of the total phosphorus and free H2PO4- accounts for ~10%. Major determinants of serum phosphorus concentration are dietary intake and gastrointestinal absorption of phosphorus, mainly via upper small intestine, urinary excretion of phosphorus, and shifts between the intracellular and extracellular spaces. Abnormalities in any of these steps can result either in hypophosphatemia or hyperphosphatemia1, 2). Lower than age-appropriate levels of serum phosphorus are associated with severe skeletal defects and growth failure, unless appropriately treated3, 4). The kidney is a major regulator of Pi homeostasis by reabsorptive capacity. Renal Pi excretion is the balance between free glomerular filtration and regulated tubular reabsorption. Under normal physiological conditions, 80-90% of filtered phosphorus is reabsorbed and the rest is excreted in the urine. Renal tubular reabsorption occurs primarily in proximal tubules by way of a transmembrane Na+ gradient-dependent process (Na+/Pi cotransport) located on the apical brush border membrane5). Most of the hormonal and metabolic factors that regulate renal tubular Pi reabsorption, including alterations in dietary phosphate content and parathyroid hormone, have been shown to modulate the proximal tubular membrane expression of the type II Na+/Pi cotransporter protein1, 6). Molecular and biochemical features of clinical disorders associated with abnormal Pi handling led to the identification of several genes and proteins involved in the maintenance of the Pi homeostasis.
Renal tubular phosphate reabsorption
1. Cellular mechanism
Renal Pi reabsorption occurs in the proximal tubule and involves the transport of Pi from the tubular lumen across the apical brush-border membrane (BBM). And then Pi absorbed by BBM Na+/Pi cotransporters leaves the cell via the basolateral transport pathway. Na+-dependent and Na+-gradient (outside>inside) mechanism is maintained by the Na+,K+-ATPase pump on the basolateral membrane.
2. Phosphate transport molecules
Three types of Na+/Pi cotransporters (types I-III; solute carrier series SLC17, SLC34, and SLC20, respectively, in the human gene nomenclature database) have been identiied in the proximal tubules of the rat kidney3,4). The type I Na+/Pi transporter is expressed in the liver and kidney3). Its expression and activity are not regulated by the dietary phosphate or PTH status. Recent studies suggest that expression of the type I gene (Npt1) is transcriptionally regulated7) and that Npt1 may function as a modulator of intrinsic cellular Pi transport rather than a Na+/Pi cotransporter8), but its role in the regulation of Pi homeostasis remains unclear9). By contrast, the type II Na+/Pi cotransporter (NPT2, NaPi2, NaPi3) is the major molecule in the renal proximal tubule and is regulated by Pi, parathyroid hormone, fibroblast growth factor 23 (FGF23) (except Type IIb), and by 1,25-dihydroxyvitamin D (1,25(OH)2D) and it is responsible for most of Pi reabsorption in the kidney and intestine5, 10, 11). Recently, three highly homologous isoforms of NPT2 have been identified. NPT2a (Type IIa) is mainly expressed in the kidney. The type IIa Na+/Pi transporter (SLC34A1) is a key mediator of Pi reabsorption in the renal proximal tubules and is affected by various hormones. The type IIa and type IIc Na+/Pi transporter is located in the apical membranes of renal proximal tubular cells3). Beck et al.12)demonstrated that disruption of the Npt2a gene in mice (Npt2a-/- mice) leads to increased urinary Pi excretion and to a 70-80% reduction in luminal BBM Na+-dependent Pi transport, which then results in hypophosphatemia. Type IIb Na+/Pi cotransporter, which exhibits wide tissue distribution and is not expressed in the kidney, is likely responsible for intestinal absorption of Pi13). Type IIc Na+/Pi cotransporter is identified as the growth-related Pi transporter expressed in the kidney14). Recent studies have led to the identification of homozygous or compound heterozygous mutations in SLC34A3, the gene encoding the Na+/Pi cotransporter NPT2c, in patients affected by HHRH (hereditary hypophosphatemic rickets with hypercalciuria)15-17). These findings indicate that NPT2c has a more important role in phosphate homeostasis than previously thought. Regulation of the type IIc Na+/Pi transporter by PTH and dietary phosphorus resembles that of the type IIa Na+/Pi transporter. Increases in the expression of type IIa Na+/Pi transporter results from hypophosphatemia quickly however, type IIc appears to act more slowly. Type III Na+/Pi transporters have been identified and show a low homology with other Na+/Pi cotransporters18, 19). These proteins have been known as receptors for gibbon ape leukemia virus (Glvr) and murine amphotropic retrovirus (Ram-1)19). In contrast to type I and type II Na+/Pi cotransporters, type III Na+/Pi cotransporters (PiT1 and PiT2) are ubiquitously expressed in most species and particularly abundant in the kidney, liver, lung, muscle, heart, and brain19). Furthermore, PiT1 and PiT2 function as Na+-dependent Pi transporters19). PiT is involved in the regulation of bone mineralization. In the kidney, type III Na+/Pi cotransporters are responsible for basolateral Pi influx in all tubular cells. Furthermore, studies suggest that elevated Pi stimulates smooth muscle cell phenotypic transition and mineralization via the activity of the type III Na+/Pi cotransporters18). Thus, the type III transporters are likely to serve as a housekeeping function and act as important mediators of cell-mediated matrix mineralization.
3. physiological regulation
Physiological regulation of Pi reabsorption involves, at the molecular level, an altered expression of a brush-border Na+/Pi cotransporter protein (type IIa Na+/Pi cotransporter)1). PTH, vitamin D, and dietary Pi intake have long been known as major regulators of serum phosphorus5). In the proximal tubules, PTH inhibits reabsorption of phosphorus via effects on NPT2a and NPT2c11, 20, 21). In the proximal tubule, PTH also acts as an inducer of mRNA encoding 25-hydroxyvitamin D-1α-hydroxylase , resulting in the formation of 1,25(OH)2D. Proximal tubular biosynthesis of 1,25(OH)2D is also induced by low serum phosphorus. Circulating 1,25(OH)2D enhances the intestinal absorption of calcium and, to a lesser extent, phosphorus. It also suppresses the biosynthesis and secretion of PTH and stimulates FGF23 synthesis. Vitamin D is suggested to increase/stimulate proximal tubular Pi reabsorption. 1,25(OH)2D treatment of rats was found to stimulate BBM Na+/Pi cotransport22). In recent studies, not only Npt2a but also Npt2c are concerned in Pi regulation. PTH and high Pi intake inhibit Na+/Pi cotransport across the BBM by altered expression of Npt2a and Npt2c proteins from the BBM to the subapical compartment. On the other hand, low dietary Pi intake and removal of PTH (parathyroidectomy) lead to an increase in BBM Na+/Pi cotransport11). FGF23, a novel regulator of renal Pi handling, inhibits both types IIa- and IIc-mediated Na+/Pi cotransport20). Various hormonal and non-hormonal factors control proximal tubular Pi reabsorption by stimulation or inhibition of BBM Na+/Pi cotransport1).
Novel factors regulating Pi homeostasis
1. PHEX
PHEX (Phosphate regulating gene with homologies to Endopeptidase, on the X chromosome) is profusely expressed on the surface of bone and teeth. The bone expression is localized to osteoblast, osteocyte, and odontoblasts. PHEX gene expression occurs in vitro and in vivo during osteoblast differentiation, and loss of PHEX function results in defective mineralization22). PHEX also plays a major role in renal phosphate handling but is not expressed in the kidney, suggesting the secondary involvement of a circulating systemic factor. Recent studies confirm that, under normal conditions, PHEX gene expression degrades and inactivates hormone-like substances. The "circulating factor" called phosphatonins promotes phosphate excretion and impairs bone mineralization. Therefore PHEX may also play a key role in phosphate homeostasis and mineralization23). PHEX gene mutations lead to underexpression of the Na+/Pi cotransporter in the kidney24). In patients with X-linked hypophosphatemia (XLH), inactivating mutations of PHEX probably result in a failure to inactivate the phosphatonins.
2. FGF23
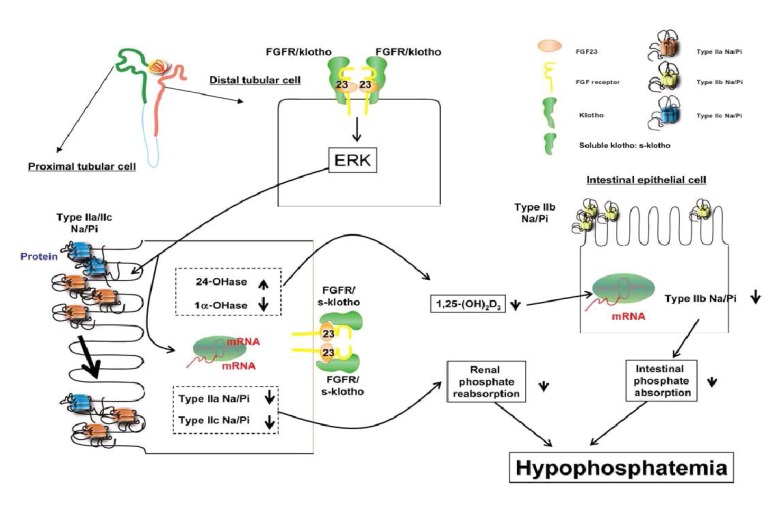
FGF23 is a recently identified member of the fibroblast growth factor family. FGF23 is thought to be one of the key molecules involved in the regulation of phosphate homeostasis and skeletogenesis25). FGF23 is required for normal phosphate balance and acts by suppressing both the reabsorption of phosphate in the renal tubule and the biosynthesis of 1,25(OH)2D. In human studies, and particularly in rodents, changes in serum phosphorus levels have been found to regulate serum FGF2326-28). FGF23 causes hypophosphatemia when injected into mice, and mice with ablation of the FGF23 gene have hyperphosphatemia and high levels of 1,25(OH)2D29). Furthermore, injection of FGF23 in mice decreases NPT2a levels and suppression of 1α-hydoxylase30). Excess circulating FGF23 concentration leads to marked depression in proximal renal tubular reabsorption of Pi. Recent studies have showed regulatory feedback mechanisms that involve the old and new regulators of phosphate homeostasis. It has been shown that 1,25(OH)2D acts as a positive regulator of FGF23 expression in bone, as demonstrated by both in vivo and in vitro studies31). FGF23 expression in bone is normally suppressed by PHEX. So deficiency of PHEX results in increased serum FGF23 and renal phosphate wasting (as seen in patients with XLH). FGF23 also inhibits PTH synthesis in the parathyroid32). Recent studies suggest that FGF23 acts via known FGF receptor (FGFR). In cultured opossum kidney cells, a cell line with a proximal tubular phenotype, FGF23 binds to the FGFR type 3c33). Klotho, a membrane bound protein with β-glucuronidase activity, is also require d as a co-receptor for FGF23 action. Klotho can bind FGF23, and its co-expression in cells converts FGFR1(IIIc) into a functional FGF23 receptor34). The Klotho null animals show markedly elevated serum levels of FGF2334). Fig. 1 shows the possible mode of action of FGF23/klotho in producing hypophosphatemia. First, FGF23 is bound to the membrane klotho/FGFR complex in the distal tubular cells or to the soluble klotho/FGFR complex in the proximal tubular cells of the kidney. Such interaction activates extracellular signal-regulated kinase (ERK) and its signaling to suppress the expression of type IIa/IIc Na+/Pi transporters in the BBM of proximal tubular cells. Alternatively, FGF23 could reduce the serum 1,25(OH)2D3 levels by suppression of 1α-hydoxylase. Reduction of the 1,25(OH)2D3 levels would result from a decrease in intestinal type IIb Na+/Pi transporter and also in a decrease in intestinal Pi absorption. The FGF23/klotho/FGFR signaling could cause hypophosphatemia by suppressing both intestinal Pi absorption and renal Pi reabsorption.
3. Other phosphaturic factors
A number of recent studies suggest that secreted frizzle-related protein 4 (SFRP4) and matrix extracellular phosphoglycoprotein (MEPE) may increase urinary phosphate excretion. Genetic studies of tumors inducing osteomalacia showed a high level of expression of the RNA for SFRP435) and MEPE36). SFRP4 on opossum kidney epithelial cells have a reduction effect in phosphate reabsorption independent from PTH. The MEPE expression was reduced by 1,25(OH)2D337) in HYP mice, a model of XLH characterized by a high level of MEPE expression.
4. Sodium-hydrogen exchanger regulatory factor 1: New renal Pi-transporter regulatory protein
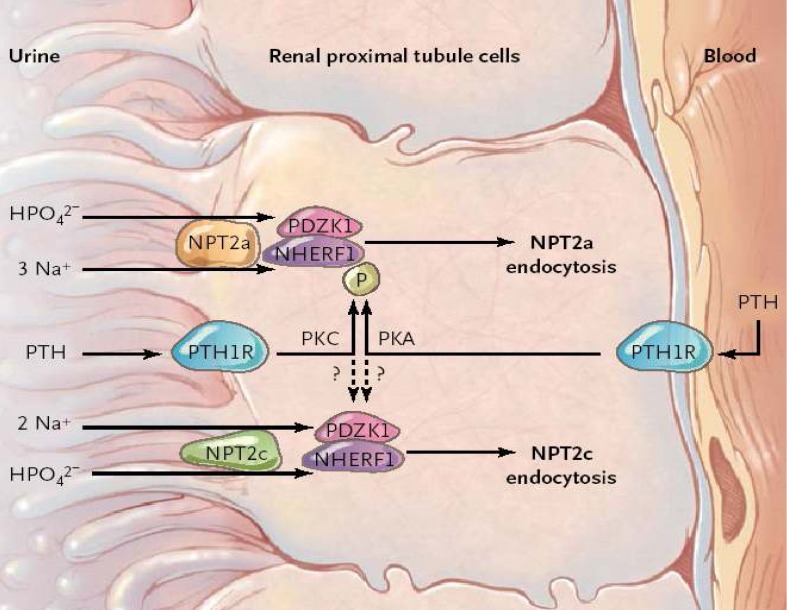
A recent study reported by Karim et al. presented another potential new mechanism of renal phosphate wasting: mutations in the sodium-hydrogen exchanger regulatory factor 1 (NHERF1)38). In the NHERF1 protein, two structural domains, named PDZ1 and PDZ2, were reported to be interacting proteins. PDZ1-domain protein interacts with the C-terminal tail of NPT2a38) and also NPT2c40) and plays an important role in renal Pi reabsorption by NPT2a41). Fig. 2 shows mechanisms of phosphorylation of NHERF1 by PTH signaling through the PTH type 1 receptor (PTH1R). Phosphorylation of NHERF1 leads to disassociation of NHERF1-NPT2a complexes, endocytosis of apical NPT2a protein, and inhibition of phosphate transport. The mechanisms of interactions between the PTH and PDZ domain containing 1 protein (PDZK1) and of PTH-induced NPT2c endocytosis remain unknown.
Inherited and aquired renal phosphate wasting disorders
1. X-linked hypophosphatemia
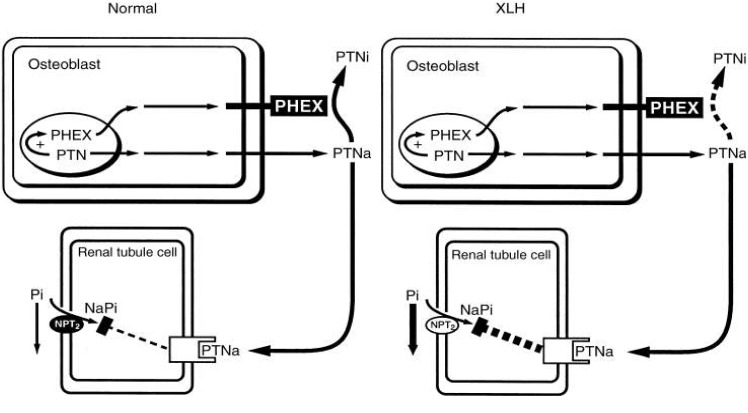
The most common inherited phosphate-wasting disorder, XLH, frequently becomes manifest during late infancy. The patient demonstrates skeletal deformities that include bowing of the long bones and widening of the metaphyseal region. These deformities are accompanied by diminished growth velocity, often resulting in short stature. In the adult stage, the patients can show osteomalacia, enthesopathy, degenerative joint disease, and continued dental disease. Hypophosphatemia in XLH patients is associated with inability of the renal proximal tubule to reabsorb phosphate. Despite the low serum phosphorus, serum 1,25(OH)2D is not elevated. Serum calcium and PTH are typically normal, although some elevation of serum PTH is observed. Genetic linkage analysis of XLH homologies and following genomic studies have demonstrated inactivating mutations in PHEX, a gene located on Xp22.142,43), since inactivating mutations lead to phosphate wasting by proteolytic cleavage failure of phosphatonin (PTN). However, PHEX-dependent proteolytic cleavage of FGF23 could not yet be demonstrated in vivo. Also, FGF23 cleavage in vitro was shown only in a single study and this could not be confirmed in others44). At present, the physiological basis of PHEX remains unknown. Under normal conditions, the osteoblast produces PHEX and PTN. The PHEX protein degrades a large amount of the active phosphatonin (PTNa) to an inactive metabolite (PTNi). The remaining circulating active hormone interacts with a renal tubule cell receptor that, by unknown mechanisms and to a small degree, down-regulates the NPT2, thereby minimally compromising the transport of phosphate. In XLH, defective PHEX fails to inactivate the majority of PTNa. Thus, excessive PTNa interacts with the renal receptor and markedly decreases NPT2 mRNA and protein content (Fig. 3)45).
2. Tumor-induced osteomalacia
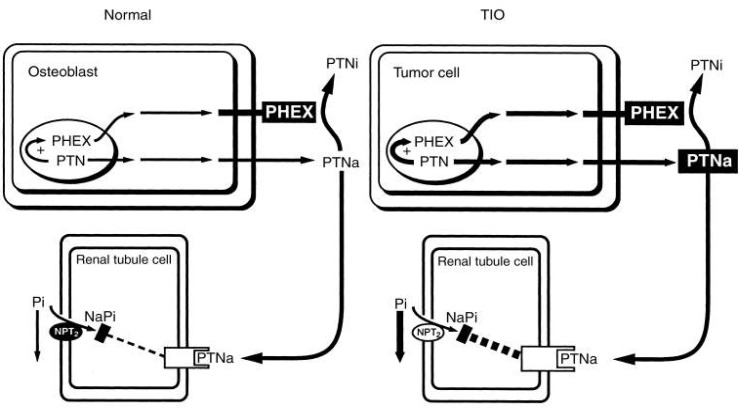
Severe hypophosphatemia with osteomalacia and, if growth plates are still open, rickets, can occur as an acquired disorder in association with a tumor. Tumor extracts inhibit phosphate transport in renal epithelial cells and reduced both phosphate and calcitriol production in experimental animals. The tumor extract affects only phosphate transport, in contrast to PTH, it has no effect on calcium metabolism. Three potential phosphaturic hormones have been concerned in Tumor-induced osteomalacia (TIO): FGF23, MEPE, and SFRP435, 36, 46, 47). Serum FGF23 was elevated in patients with TIO and fell after surgery for removal. Clinical features are similar to XLH. Plasma calcitriol level is reduced, even though elevated levels are to be expected in the presence of hypophosphatemia. Thus, the underlying tubular defect that impairs phosphate reabsorption also appears to affect calcitriol synthesis. TIO tumor cells produce PTNa in excess. The increased PTN production, through a feedback mechanism, enhances PHEX production. However, the overproduction of PTNa exceeds the capability of PHEX to degrade sufficient amounts of the product to PTNi. Hence, in spite of enhanced PHEX, with an overabundance of PTNa, interaction with the receptor decreases the NPT2 mRNA and protein production (Fig. 4)45).
3. Type IIa Na+/Pi cotransporter deficiency
The homozygous ablation of Npt2a gene in mice (Npt2a-/-) results from increased urinary phosphate excretion leading to hypophosphatemia12). Npt2c protein abundance is significantly increased in Npt2a-/- mice48), although up-regulation of Npt2c is not sufficient to compensate for loss of Npt2a function. Due to the hypophosphatemia, Npt2a-ablated mice show an appropriate elevation in the serum levels of 1,25(OH)2D leading to hypercalcemia, hypercalciuria, and decreased serum PTH levels. A study reported by Prie D et al.49) showed that heterozygous mutations in the NPT2a gene may be responsible for hypophosphatemia and urinary phosphate loss in patients with urolithiasis or bone demineralization.
4. NHERF1 mutations
Recent studies from animal models suggest that NHERF1 controls renal phosphate transport. The study reported by Karim et al.38) identifies NHERF1 mutations as a cause of renal phosphate loss that may increase the risk of renal stone formation or bone demineralization together with normal serum PTH concentrations. This study was carried out for the NHERF1 gene in 158 patients, 94 of whom had either nephrolithiasis or bone demineralization and identified three distinct mutations in seven patients with a low value of tubular maximal reabsorption of phosphate corrected for glomerular filtration rate (TmP/GFR). This study also showed increased PTH-induced cyclic adenosine monophosphate (cAMP) generation and then the inhibition of phosphate transport. Urinary cAMP excretion was significantly higher in the patients with NHERF1 mutations than patients without NHERF1 mutations38). PTH induced a significant decrease of phosphate uptake in all cell groups38). However, both PTH-induced cAMP generation and PTH-induced inhibition of phosphate uptake were increased in mutant NHERF1 complementary DNA (cDNA) as compared with human wild-type NHERF1 cDNA38).
5. Autosomal dominant hypophosphatemic rickets/osteomalacia
Autosomal dominant hypophophatemic rickets (ADHR) is a rare isolated renal phosphate wasting disease with rickets or osteomalacia that is transmitted as an autosomal dominant trait. ADHR results from heterozygous mutations in FGF23 gene on chromosome 12p1350). In ADHR circulating FGF23 increased because PHEX cannot inactivate the mutated form of FGF23. Clinical manifestations are similar to X-linked disease but exihibits severe natured manifestations. Inappropriately low or normal 1,25(OH)2D levels are observed in patients with ADHR.
6. Hereditary hypophosphatemic rickets with hypercalciuria
HHRH is autosomal recessive genetic disorder caused by mutations of the renal type IIc Na+/Pi cotransporter, which contains the gene SLC34A3 in chromosome 9q3415-17). Hypophosphatemic rickets and/or osteomalacia is the clinical manifestation in most patients. Nephrolithiasis associated with hypercalciuria frequently occurs, probably due to elevated serum 1,25(OH)2D that leads to increased intestinal absorption of calcium and phosphorus. Serum FGF23 is low to low-normal in HHRH15). Long-term phosphate supplementation is the only therapy in HHRH.
Conclusion
PTH and 1,25(OH)2D have been investigated as the most important regulators of phosphate homeostasis. FGF23 and PHEX are novel renal Pi regulator proteins, which are mutated in ADHR and XLH respectively. PHEX is an important negative regulator of FGF23. PTH and FGF23 both inhibit proximal tubular phosphate reabsorption. However, whereas PTH stimulates the synthesis of 1,25(OH)2D, FGF23 inhibits this. FGF23 appears to act via known FGFRs, but Klotho protein, as a co-receptor, is required for the action of FGF23. Mutations in the genes encoding two renal Na+/Pi transporters, NPT2a and NPT2c, have been identified in patients with acquired and genetic Pi wasting disorders. In recent studies, NHERF1 was reported as another new regulator for the Pi reabsorption mechanism. NHERF1 phosphorylation by PTH has been shown to be important in the endocytosis of NPT2a. In humans, NHERF1 mutations play a causative role in patients with unexplained hypophosphatemia. Investigations for various phosphaturic hormones FGF23, SFRP4, MEPE, etc and renal phosphate transporter genesare underway to define their mechanism on renal Pi regulation.
 XML Download
XML Download