

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Despite the presence of similar degree of severe hyperglycemia in both nonketotic hyperosmolar status (NKHS) and hyperglycemia on maintenance dialysis (HMD), the absence of osmotic diuresis due to the negligible renal function in patients with HMD was assumed to cause the different clinical manifestations from those of NKHS with intact renal function in general1-3).
Clinical pictures are related mostly to severe dehydration or hypovolemia due to the profound losses of water and solutes in NKHS. The most common presenting manifestations were dyspnea (51%) and pulmonary edema (44%) followed by nausea and vomiting (35%) and even no symptoms at all (12%) in a recent analysis of 43 episodes of HMD3).
Hereby, we report a case of HMD admitted due to recurrent symptomatic hyperglycemia always with nausea and vomiting. However, these symptoms were found to be related to the gastroparesis with noncompliance to prokinetic medications rather than to the hyperosmolality due to hyperglycemia itself.
Case report
On March 15, 2007, the 60 year-old-male on maintenance hemodialysis for the past 2 years with a known history of type 2 diabetes mellitus for 22 years, and hypertension for 15 years, was admitted via emergency room due to hyperglycemia accompanied by severe nausea and vomiting for 3 days prior to admission. Also, he had a history of diabetic gastroparesis for the past 3 years, which was diagnosed on the basis of clinical symptoms of nausea and vomiting with postprandial abdominal bloating, the documented delayed gastric emptying time in gastrointestinal motility test, and significant improvement after initiation of prokinetic agents on initial admission.
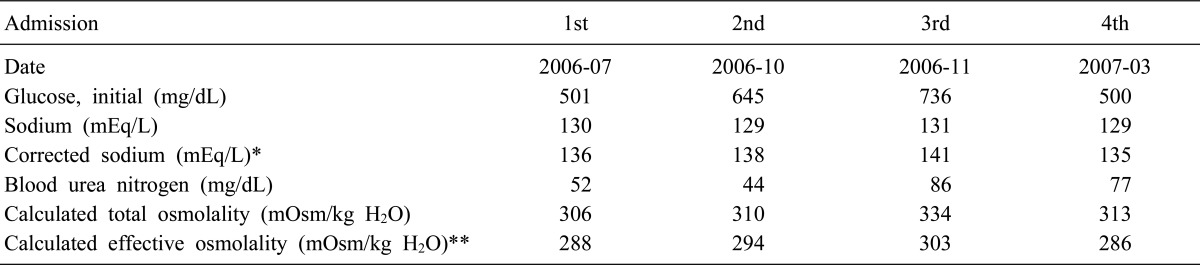
He was admitted 3 times within 1 year before this admission with recurrent hyperglycemia and same symptoms, nausea and vomiting, as shown in Table 1. Upon each admission, hyperglycemia was well corrected with regular insulin infusion within 24 to 48 hours. Also, the accompanied symptom of nausea and vomiting dissipated simultaneously. Therefore, the diagnosis on discharge was HMD, and nausea and vomiting were assumed due to the presenting symptoms of HMD3). Previous medication has been prescribed water soluble vitamins, calcium-carbonate, irbesartan (150 mg/day), nateglinide (240 mg/day) as oral hypoglycemic agent, metoclopramide (15 mg/day) and mosapride (15 mg/day) with each meal as prokinetics, and erythropoietin (8,000 units/week, subcutaneous).
In the emergency room, his blood pressure was 160/90 mmHg and other vital signs were stable. He was alert and oriented. His tongue was mildly dehydrated and also skin turgor was mildly poor. Abdomen was soft with no tenderness, and his bowel sounds were present. Laboratory findings were as follows: white blood cells 10,700/mm3, hemoglobin 10.5 g/dL, hematocrit 31.4%, platelet 414,000/mm3, serum sodium 129 mEq/L, serum potassium 4.9 mEq/L, serum chloride 82 mEq/L, total protein 8.4 g/dL, serum albumin 4.7 g/dL, serum aspartate aminotransferase/alanine aminotransferase 15/9 U/L, serum calcium 10.1 mg/dL, serum phosphorus 7.8 mg/dL, blood urea nitrogen (BUN) 77 mg/dL, serum creatinine 8.7 mg/dL, and serum glucose 500 mg/dL. The corrected serum sodium by the method of Katz4), which assesses that serum sodium concentration falls by 1.6 mEq/L for every 100 mg/dL increment in serum glucose value, was 135 mEq/L. The calculated total serum osmolality to the formula, 2[Na, mEq/L]+[glucose, mg/dL]/18+[BUN, mg/dL]/2.8, was 313 mOsm/kg H2O5). However, the calculated effective serum osmolality, i.e., hypertonicity, by subtracting the osmolality due to urea from total serum osmolality, was only 286 mOsm/kg H2O (Table 1)6).
As on his previous admissions, the initial impression was HMD. His blood sugar was well controlled with 24 units of regular insulin in total during the initial 48 hrs following his latest admission, however, nausea and vomiting was persistent with anorexia for the next 5 days. Esophagogastroscopy revealed linear erosion over his middle and lower esophagus with retained food material even after fasting overnight, which was consistent with reflux esophagitis due to severe gastroparesis. These clinical findings required doubling the dose of two kinds of prokinetics for gastroparesis, thereafter, persistent nausea and vomiting disappeared gradually but completely. He admitted the omission of medication (prokinetics and oral hypoglycemic agent) for uncertain reasons for at least 2 to 3 days before each previous admission to the hospital. Therefore, he was repeatedly educated for the importance of regular intake of doubled dose of prokinetics with intake of small volume of meals, and then was discharged on day 9. Following the discharge, he has been under the supervision of dialysis personnels for strict compliance to medication, especially for prokinetics, when visiting for regular hemodialysis for 3 times a week. He has been remaining symptom-free and in euglycemic status without further admission for the past 5 months.
Discussion
In the present case the repeated admissions showed similar clinical characteristics of HMD as described in 12 diabetic patients on dialysis by Al-Kudsi et al as follows2): 1) alert and oriented mental status, 2) no distinct signs of dehydration or hypovolemia, 3) nausea and vomiting as the main chief complaint, and 4) successful correction of hyperglycemia with small doses of regular insulin within 48 hrs. Among these clinical characteristics, recurrent symptoms of nausea and vomiting could be related not only to the presenting clinical manifestation of the HMD but also to gastroparesis as its precipitating factor3).
Hyperglycemia in the NKHS causes coma and other neurologic deficits. Osmotic diuresis has an important role in these circumstances7). On the contrary, the absence of glycosuria with negligible renal function as our case in HMD does not develop osmotic diuresis, which may be the main reason for the paucity of neurologic symptoms as well as the other signs and symptoms of severe dehydration. However, the accumulation of glucose in the extracellular compartment in the absence of osmotic glycosuria causes both hypertonicity and shift of water from the intracellular into the extracellular compartment leading to hyponatremia.
In the analysis of data in a series of HMD (n=12) in comparison to NKHS, despite the similar degree of hyperglycemia, the serum sodium level averaged only 125 mEq/L, a value considerably less than those found in the NKHS, 144 mEq/L, and accordingly, the calculated effective osmolality (tonicity) in HMD was 316 mEq/L compared to 384 mEq/L in NKHS1, 2). Similarly, because of the low serum sodium levels ranging from 129 to 131 mEq/L, our patient's calculated effective osmolality (tonicity) ranged only from 288 to 303 mOsm/kg H2O (Table 1). Therefore, absence of osmotic diuresis and dilution of the extracellular compartment by intracellular water transfer seems to be the main causes of modest hypertonicity in HMD as was seen in case.
As described above, despite the fact that changes in serum tonicity levels caused by hyperglycemia is generally lower in HMD than in NKHS, hyperglycemia produces extracellular fluid volume changes in opposite directions in the two different states of renal function. Patients with NKHS routinely develop symptomatic extracellular volume deficit and require large volumes of fluid replacement, whereas patients with HMD on negligible renal function reveal extracellular volume expansion from the osmotic transfer of intracellular fluid into the extracellular fluid compartment. This expansion of volume in the extracellular fluid compartment can cause pulmonary edema and dyspnea, which was in fact the most common presenting symptoms in a report of HMD3). Also, this extracellular volume expansion in HMD rather than extracellular volume deficit is responsible for the easy reversibility of hyperglycemia with insulin therapy rather than volume replacement as shown in our case. Therefore, in HMD, administration of insulin alone is usually sufficient to correct fluid and hyperosmolality and the accompanied clinical manifestations.
Our patient with HMD was presented to the emergency room with recurrent symptoms of nausea and vomiting. Nausea and vomiting could be one of the gastrointestinal symptoms due to severe hyperglycemia in HMD, and it was the one of the common presenting clinical manifestations preceded by pulmonary edema and dyspnea only3). However, symptomatic diabetic gastroparesis (DGP) manifested by chronic nausea and vomiting develops in approximately 50% of diabetics on maintenance dialysis. Furthermore, uncontrolled hyperglycemia (50%) is more prevalent than hypoglycemia (10%) in DGP, and poor glycemic control seems to be its major pathogenetic factor8). In another point of view, DGP and poor glycemic control in HMD could be closely linked with undetermined relation of the cause and effect between them. However, in our patient, a case with HMD, showed evidence that uncontrolled DGP was the cause of uncontrolled hyperglycemia.
 XML Download
XML Download