

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Vasopressin, the antidiuretic hormone, is the major physiologic regulator of renal water excretion and plays an important role in regulating serum osmolality. Several clinical conditions have been associated with inappropriately increased vasopressin secretion that causes antidiuresis, with subsequent retention of water by the kidney. The impaired renal water excretion results in increasing total body water, causing hypo-osmolality and hyponatremia.
Vasopressin synthesis and regulation of secretion
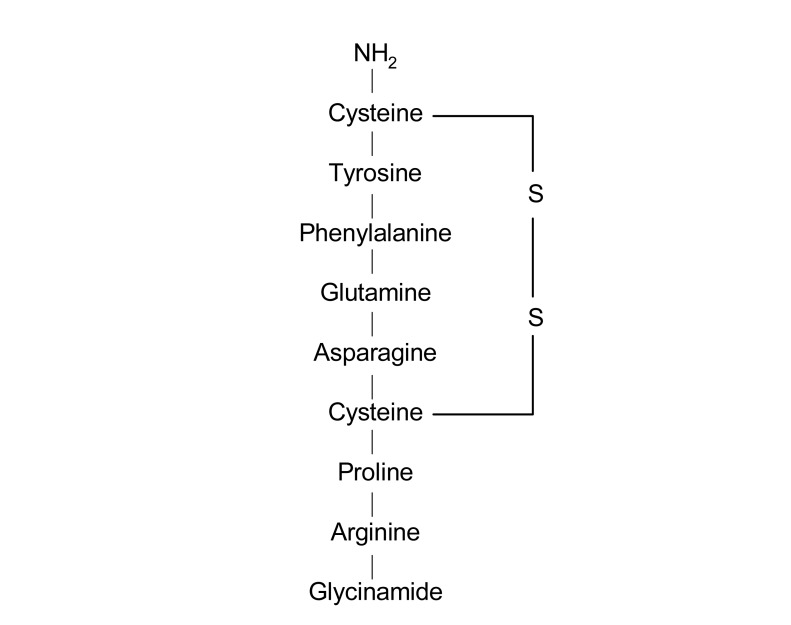
Vasopressin is a nonapeptide (molecular weight 1,099) synthesized in the paraventricular and supraoptic nuclei of the hypothalamus (Fig. 1). The hormone is transported to the posterior pituitary gland and stored. It is released to the bloodstream upon appropriate stimulation. The most important mechanism involved in the control of the release of vasopressin is the effective osmotic pressure of plasma. Changes of as little as 1% in plasma osmolality can cause significant increases in plasma vasopressin levels with proportional increases in urine concentration. Maximal antidiuresis is achieved after increases in plasma osmolality of only 2-4% above the threshold for vasopressin secretion. In addition, the rapid response of pituitary vasopressin secretion and its short half life enable adjustment of renal water excretion to changes in plasma osmolality on a minute-to-minute basis.
Hypovolemia which is detected by arterial and atrial baroreceptors is also a potent stimulus for vasopressin secretion. Although the osmotic mechanism of regulation of vasopressin secretion is very sensitive, a 15-20% fall in blood pressure is required to induce maximal antidiuresis. Vasopressin release can be influenced by a variety of other factors that are not directly related to osmolal or volume balance. Nausea, hypoglycemia, angiotensin II, nonspecific stress, and acute hypoxia or hypercapnia stimulate vasopressin secretion, whereas drinking lowers plasma vasopressin before there is any appreciable decrease in plasma osmolality. A variety of drugs nicotine, also stimulate or inhibit vasopressin secretion1).
Vasopressin receptors
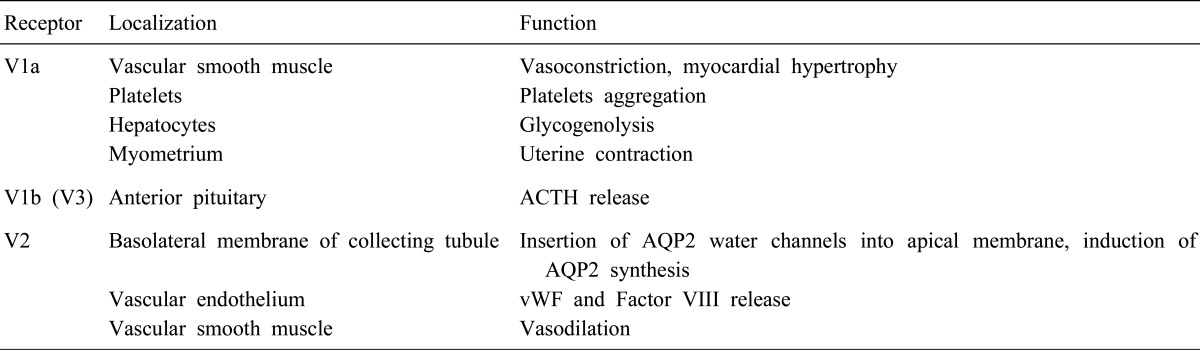
V1a, V2, and V1b (or V3) vasopressin receptors have been identified. The receptors are members of the G-protein coupled receptor superfamily. Activation of V1a receptors located in vascular smooth muscle cells, myocardium, hepatocyte, platelets, and myometrium results in vasoconstriction, myocardial hypertrophy, hepatic glycogenolysis, platelet aggregation, and uterine contraction. V1b receptors are mainly located in the anterior pituitary and regulate the secretion of adrenocorticotropic hormone (ACTH). The V2 receptors (V2R), present in the basolateral membrane of the principal cells of the renal collecting tubules and connecting tubules, mediate the osmotic effect of vasopressin2). The V2R are also present in thick ascending limbs of the loop of Henle and stimulate NaCl reabsorption into the medullary interstitium3). They are found in vascular endothelium and effect release of the von Willebrand factor and Factor VIII (Table 1).
The aquaporins
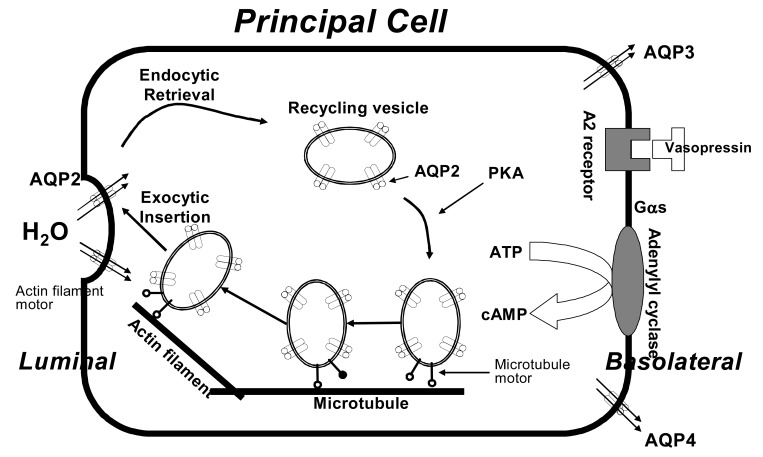
The V2R is a 41 KDa protein of 371 residues with seven transmembrane domains. It is expressed on the plasma membrane of collecting duct principal cells. Binding of vasopressin to the V2R activates the stimulatory heterotrimeric GTP-binding protein (Gs). The activated Gs then stimulates adenylate cyclase, resulting in an increase in intracellular cAMP. The latter activates protein kinase A, which phosphorylates preformed aquaporin-2 (AQP2) water channels located in intracellular vesicles. Phosphorylation of AQP2 promotes trafficking to the apical membrane, followed by exocytic insertion of AQP2 vesicles into the cell membrane. This is the rate limiting step for transepithelial water flow. Aquaporins-3 and -4 (AQP3 and AQP4) are constitutively present in the basolateral membrane. However, AQP3 is only regulated by vasopressin. AQP3 expression changes following vasopressin stimulation and several pathologic states. When the effect of vasopressin is dissipated, the water channels are removed from the apical membrane by endocytosis and returned to the cytoplasm. AQP2 expression levels markedly increase in response to long term dehydration, with an increased abundance of AQP2 in the apical plasma membrane. Therefore, collecting duct water permeability is regulated by short-term and long-term mechanisms, both critically involving AQP24, 5) (Fig. 2).
Role of vasopressin in hyponatremic states
Plasma vasopressin levels have been found to be inappropriately elevated in several conditions such as congestive heart failure (CHF), syndrome of inappropriate antidiuretic hormone secretion (SIADH), liver cirrhosis (LC), polycystic kidney disease, nephrotic syndrome, and surgical stress. In SIADH, vasopressin release is not fully suppressed despite hypotonicity. In CHF and LC, a diminished glomerular filtration rate or impaired delivery of solute to the diluting segment impair maximal water excretory capacity. However, the persistence of vasopressin release due to nonosmotic stimuli is predominantly responsible for water retention in these disorders6).
Vasopressin receptor antagonists
Vasopressin receptor antagonists (VRAs) was first developed as a peptide antagonist during the 1970s. Although a peptide mediate V2R antagonist effect in animals, a phase 1 trial showed that the agent had a paradoxical weak V2R agonist in human studies. Thus, further development of peptide antagonists was abandoned7). Using a functional screening strategy, non-peptide VRAs were subsequently identified. The four non-peptide agents, include conivaptan, lixivaptan, tolvaptan, and satavaptan, are now in various stages of clinical trials (Table 2). Molecular modeling of binding sites suggests that these antagonists penetrate deeper into the transmembrane region of the V2R than vasopressin. They thereby prevent binding of native hormone8).
Conivaptan is a combined V1aR and V2R antagonist, whereas others are selective V2R antagonists. All Agents of this class are inhibitors of the cytochrome P350 3A4 system, but conivaptan is the most potent. In December 2005, the US FDA approved conivaptan for treatment of euvolemic hyponatremia. Although the drug is orally active, to minimize the possibility of drug interaction, the FDA has restricted its distribution to a parenteral form for short-term in-hospital use only. Other VRAs are being developed for long-term oral use9).
Potential uses for vasopressin receptor antagonists
1. Hyponatremia
In many clinical trials, these agents produce an aquaresis leading to increased plasma Na+ concentration in the majority patients with hyponatremia due to SIADH, CHF, and LC. Although the FDA approved conivaptan only for the indication of euvolemic hyponatremia, increased exposure of larger numbers of clinical trials should lead to approval for the indication of hypervolemic hyponatremia. The optimum use of VRAs has not yet been determined, but some predictions can be made according to clinical trials. For hyponatremia in hospitalized patients, parenteral conivaptan will likely be the preferred agents. In patients for whom oral therapy is suitable and for chronic forms of hyponatremia, selective V2R antagonists such as lixivaptan, tolvaptan, and satavaptan will likely be useful.
Safety issues must also be considered carefully. The possibility of ovecorrection has been significantly concerned, but osmotic demyelination has not been reported in all of the clinical trials. Safeguard, such as avoidance of correction of the plasma sodium concentration at a rate faster than 8-12 mEq/L/24hr, will be sufficient to protect against overly rapid correction of plasma sodium concentration. A second concern is to avoid using VRAs in cases of hypovalemic hyponatremia, where an aquaresis would aggravate the underlying volume depletion. Hypotension, particularly with conivaptan, or bleeding complications should be carefully monitored9, 10).
2. Congestive heart failure
In the ACTIV in CHF (Acute and Chronic therapeutic Impact of a Vasopressn Antagonist in Congestive Heart Failure) trial, 319 hospitalized patients with heart failure exacerbation were randomized to tolvaptan or placebo. Worsening CHF or unscheduled visits for CHF within 60 days were not different among the treatment groups, although post-hoc analysis suggests that cardiovascular mortality may be reduced in the tolvaptan group in the higher risk group with kidney function impairment or severe congestive findings11). Until well-powered studies, to determine whether VRA therapy will have beneficial effect on the CHF neurohormonal cascade have been completed, VRAs are not recommended in patients with CHF.
3. Liver cirrhosis
Because the presence of portal hypertension in these patients, V1aR blockade may produce hypotension and variceal bleeding owing to antagonism of vasopressin pressor effects in this vascular bed. Thus, only selective V2R antagonists can be recommended for these patients9).
4. Other diseases
A potential role for the VRAs in attenuating polyuria in nephrogenic diabetes insipidus and cyst development in polycystic kidney disease is being explored9).
Conclusion
Vasopressin is a neuropeptide hormone that plays an important role in water metabolism via regulation of AQP2 in the principal cells of the renal collecting tubules and connecting tubules. The SIADH, CHF, and LC are among many conditions associated with abnormal water retention mediated by vasopressin release inappropriate to plasma osmolality. VRAs target the cause of abnormal water retention by producing an increase in electrolyte-free water excretion. Thus, these agents will provide highly specific therapy for euvolemic or hypervolemic hyponatremia, CHF, and LC.

 XML Download
XML Download