

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The systemic role of the renin-angiotensin system (RAS) in the regulation of blood pressure and volume homeostasis and in the pathophysiology of hypertension (HTN) has been targeted for many decades1). Increased RAS activity is also a major determinant for numerous pathologic conditions because angiotensin II (Ang II) increases aldosterone and blood pressure and contributes to the development of end-organ damage through direct effects on cardiac, vascular, and renal tissues2).
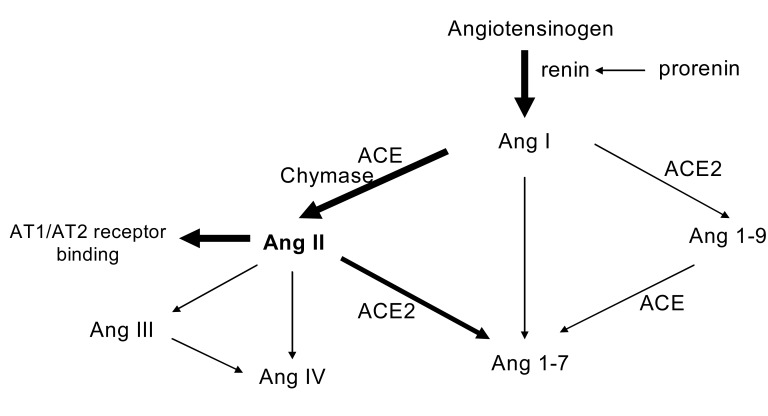
As is well known, Ang II is produced systemically. The substrate of the RAS, angiotensinogen (AGT) is released from the liver and is cleaved in the circulation by renin that is secreted from the juxtaglomerular apparatus of the kidney to form Ang I. Then, angiotension I (Ang I) is easily activated to Ang II by angiotensin converting enzyme (ACE), which is predominantly expressed in high levels on the surface of endothelial cells in the pulmonary circulation3). Ang II, considered as the most powerful active product of the RAS, acts on specific receptors. Notably, this view of the RAS has been expanded by more recent findings that increased the complexity of the system. Different Ang receptors and signal transduction pathways have been described. Moreover, additional other peptides such as Ang 1~7, have been recognized, and alternative pathways of Ang II formation, for example, by the serine protease chymase have been proposed4) (Fig. 1). The resulting alteration of our inspection of the RAS has introduced the concept of "local" or "tissue" RAS2). Among these, the pathophysiological implications of the intrarenal RAS have been the main focus of attention in the pathogenesis of HTN and progressive renal injury. A large body of data is now available to support the cardinal role of intrarenal RAS exerting diverse pathologic effects on the development and progression of HTN and renal damage.
The widespread Ang II actions at the cellular level, such as cell growth and apoptosis, can be the main factors of important physiological stimuli. Transgenic overexpression and knock-out animal models of RAS genes have demonstrated a functional role of the RAS in prenatal development5, 6). Additionally, numerous epidemiological and experimental studies have provided the profound involvement of the RAS in the fetal programming of hypertension and adult disease7, 8). Programming during fetal life occurs aligned with an adverse fetal environment and leads to long-term adaptive responses that lead to structural and physiological changes and the subsequent development of HTN9, 10).
In this review, we will summarize the physiologic actions of Ang II including the intrarenal RAS and the recent findings associated with its roles in the pathogenesis of HTN. We will also discuss evidence regarding the effects of the intrarenal RAS on fetal programming of HTN.
Physiologic effects of angiotensin II
The critical physiologic actions of the RAS are the management of blood pressure stability and extracellular fluid volume homeostasis. Most of these actions of the RAS are executed by Ang II with its receptor in a variety of organs and tissues. Ang II is one of the most powerful vasoconstrictors known and acts on the heart, vasculature, nervous system, digestive organs, skin, reproductive tract, sensory organs, lymphatic tissue, adipose tissue, adrenals, and kidneys. It appears likely that local and systemic actions of the RAS have to be integrated in a concerted action of Ang II-mediated effects2).
In the kidney, Ang II participates in vascular, tubular, and growth-promoting activities. Administration of exogenous Ang II decreases renal blood flow and glomerular filtration rate (GFR), and constricts afferent and efferent arterioles dose-dependently11). Afferent arteriole vasoconstrictor responses to Ang II are mediated by Ang II type 1A (AT1A) and 1B (AT1B) receptors, whereas efferent arteriolar vasoconstrictor responses to Ang II are mediated by only AT1A receptors in the mouse kidney12). Ang II also reduces the glomerular filtration coefficient while increasing afferent and efferent arteriolar resistances, which contributes to the decreases in GFR.
Acute Ang II infusion sufficient to change renal hemodynamics does not cause proteinuria. However, sustained elevation of intrarenal Ang II induces proteinuria accompanied by progressive injury of the glomerular filtration barrier, which is composed of the glomerular endothelium, glomerular basement membrane, and podocytes13).
The effect of pharmacologic blockade of Ang II on GFR is quite variable, either increased, decreased, or unchanged. Most clinical studies show that the GFR remains stable when Ang II blockade is introduced. When the arterial pressure is not obviously reduced, Ang II blockade increases the single nephron GFR as well as the single nephron plasma flow. In contrast, a significant reduction in the GFR has often been seen in patients with renal disease. Presumably, blood pressure reduction by Ang II blockade stimulates sympathetic nerve activity, which prevents afferent arteriolar vasodilation, allowing glomerular pressure to fall1). It may also be difficult to maintain the GFR by sufficient increases in the glomerular filtration coefficient when glomerular pressure is reduced by treatment with ACE inhibitors and AT1 receptor blockers (ARBs)14).
Ang II also exerts a modulatory effect on the sensitivity of the tubuloglomerular feedback mechanism, which provides a balance between the reabsorption of the tubules and the filtered load by adjusting the GFR. Micropuncture analysis in transgenic mice showed an essential role of Ang II in tubuloglomerular feedback regulation mediated through the AT1A receptor15).
Ang II is involved in the regulation of renal sodium and water excretion in not only via effects on renal hemodynamics, GRF, and regulation of aldosterone secretion, but also via direct effects on renal tubule transport. It also stimulates H+ secretion and HCO3- reabsorption in both proximal and distal tubules and regulates H+-ATPase activity in the intercalated cells of the collecting tubule16). The activation of apical Na+/H+ exchange, basolateral Na+/HCO3- cotransport, and basolateral Na+,K+-ATPase and apical H+-ATPase are implicated in Ang II inducedtranscellular sodium and bicarbonate reabsorption within the proximal tubule, whereas Na+/H+ exchange and H+-ATPase contribute to the reabsorption of sodium and bicarbonate in distal tubules. Furthermore, abnormal water metabolism in mice lacking the AT1A receptor has been shown17), suggesting that Ang II stimulates the urinary concentrating mechanism in inner medullary collecting ducts, leading to increased water reabsorption.
Substantial data provides compelling evidence that Ang II has growth effects for renal cells. Whether Ang II either mediates proliferation or rather hypertrophy depends on the renal cell type. All components of the RAS are developmentally regulated in renal organogenesis, and blocking the RAS in the developing kidney shows severe renal abnormalities and abnormal cellular turnover18). Ang II-induced growth effects are also implicated in the development of glomerulosclerosis and tubulointerstitial fibrosis and antagonizing these effects is a primary target of renoprotection modality in clinical nephrology.
Intrarenal renin-angiotensin system
Though every organ system has components of the RAS, the kidney is exclusive in having every element of the RAS with compartmentalization in the tubular and interstitial networks as well as intracellular accumulation14). Not surprisingly, the key role of tissue as well as classical RAS in blood pressure regulation has well been recognized.
1. Angiotensinogen
Human AGT is expressed in multiple tissues, including liver, adipose tissue, heart, vessel wall, brain, and kidney. The human AGT glycoprotein contains a signal peptide that is removed cotranslationally to yield the substrate of renin. Whereas AGT is constitutively secreted, recent evidence suggests that the protein may localize in the nucleus of some cells. Intrarenal AGT mRNA and protein are localized to proximal tubule cells, suggesting that the intratubular Ang II could be derived from locally formed and secreted AGT19). The proximally formed AGT is screted into the tubular fluid and flows into the distal nephron, allowing intraluminal Ang II formation to continue throughout the nephron with the residual AGT appearing in the urine.
Plasma AGT levels are close enough to the Michaelis constant for renin that small increases in either renin or AGT may increase Ang II production and alter blood pressure14). Notably, elevated renal-specific expression of AGT causes systemic hypertension without a change in circulating Ang II. AGT expression may exert their effects in a tissue-specific manner and not necessarily by changing plasma AGT. Transgenic mice overexpressing rat AGT gene in the kidney significantly increased blood pressure, albuminuria, and renal injury; and administration of ARB or ACE inhibitor reversed these abnormalities20).
2. Renin and prorenin
Renin is an aspartyl-protease that exists in two forms, the proenzyme prorenin and mature renin. The secreted mature renin has 339 to 343 amino acid residues after proteolytic removal of the 43-amino acid residue at the N terminus of prorenin. Myo-epithelioid cells of the juxtaglomerular apparatus are involved in the synthesis and the processing of prorenin into mature renin and the release of renin in the circulation. Positive renin expression has been observed in cells of glomeruli, proximal and distal nephron, and connecting tubules. Renin activity was also observed in excreted urine. The release of both renin and prorenin is thought to occur in response to a reduction in glomerular afferent arteriolar pressure, sympathetic nerve stimulation, or a reduced rate of sodium delivery to the distal tubules.
In the kidney, the recently described renin receptor binds renin and prorenin, leading to an increase in the catalytic efficiency of Ang I formation from AGT21). The direct renin inhibitor may be a more acceptable access to inhibiting the RAS as it inhibits renin binding to AGT, which is ultimately the rate-limiting step in Ang II production. There is also a supporting evidence that the binding of renin to the receptor can have profibrotic effects that are independent of Ang II, suggesting direct actions of renin and prorenin on organ pathophysiology22). Renin itself or prorenin may directly affect cellular effects.
3. Angiotensin converting enzyme
ACE is found in many vascular endothelial cells and in membranes of various other cells including the brush border membranes of the proximal tubules. ACE operates as a "peptidyl dipeptidase" removing dipeptides from the C terminus of peptide substrates. Its primary substrate is Ang I and it processes the decapeptide Ang I to the 8-amino acid peptide Ang II. The ACE plays pivotal roles in the regulation of cardiovascular function and fluid and electrolyte homeostasis. ACE also degrades bradykinin to inactive fragments, reducing the serum level of endogenous vasodilators. The localization of ACE within the kidney is different between humans and commonly used experimental animals. Human kidneys predominantly expressed ACE in the brush border of proximal tubules, whereas rat kidneys intensely presented it on the endothelial cells of the renal microvasculature23).
4. Angiotensin II receptor
In various regions, nephron segments, and cell types within the kidney, Ang II receptors play a key role in the complex and extensive actions of Ang II on renal function. AT1 (subtypes 1A and 1B) and Ang II type 2 (AT2) receptors are two major types of Ang II receptors. AT1 receptor mainly contributes to most of the Ang II actions and has been widely distributed throughout the kidney. In Ang II-dependent hypertension, vascular and glomerular AT1 receptors are down-regulated, but the proximal tubular receptors are either up-regulated or not altered24). AT1A receptor-deficient mice showed a predominant role of AT1A receptors and a limited role of AT1B receptors in blood pressure regulation and in the renal responses to long-term Ang II administration25).
AT2 receptor expression is found in proximal tubules, collecting ducts, glomerular epithelial cells, and some of the vasculature. The AT2 receptor increases highly during fetal life and decreases dramatically after birth. It is supposed that the actions mediated via the AT2 receptor generally oppose those carried out by the AT1 receptor, although the functions of the AT2 receptor are largely unknown. Absence of the AT2 receptor leads to vascular and renal hypersensitivity to Ang II, including sustained antinatriuresis and hypertension, suggesting that the AT2 receptor plays a counterregulatory protective role mediated via bradykinin and nitric oxide against the antinatriuretic and pressor actions of Ang II26).
5. Intrarenal angiotensin II
Within the cortex, Ang II is distributed in the interstitial fluid, tubular fluid, and intracellular compartments. Ang II concentrations in interstitial fluid are much higher than the plasma levels in several experimental models of Ang II-dependent hypertension, indicating substantial local formation. Proximal tubule fluid levels of Ang I and Ang II are also known to be much greater than the plasma concentrations, however, the Ang II levels in other tubular fluid remain to be established. Several studies support an important role for Ang II in regulating reabsorptive function in the distal nephron and collecting duct segments, as well as in proximal tubule segments, which activate the Ang II receptors on the luminal borders27).
Representative data also shows some of the Ang II that binds to receptors is internalized via AT1 receptor-mediated endocytosis. A higher fraction of intrarenal Ang II is internalized into intracellular endosomes via an AT1 receptor mediated process in Ang II-dependent hypertension. Endocytosis of the Ang II-AT1 receptor complex seems to be necessary for the full expression of functional responses coupled to the activation of signal transduction pathways, and the internalized Ang II could be recycled and secreted to exert further genomic actions by binding to Ang II receptors on the cell membranes, cytosol, and nucleus28).
6. Alternative pathway
Increasing evidence has shown that alternative pathways to the ACE exist for the Ang II generation in the heart, arteries, and kidney. Of these, the chymase-dependent pathway is thought to be the most important, as Ang II formation is substantially blocked by chymase inhibitors. More than 80% of Ang II formation in the human heart and more than 60% of that in arteries seems to be chymase-dependent, and recent studies support the potential contribution of chymase-dependent intrarenal Ang II formation to the progression of renal injury. ACE-knockout mice have shown that local Ang II generation within the kidney is unchanged due to a 14-fold increase in the chymase activity29) and intra-arterial infusion of a chymase inhibitor significantly diminished intrarenal Ang II in the ischemic kidney30). Clinical studies reported increased chymase expression in rejected kidneys and kidneys of patients with renovascular hypertension and diabetes.
In the classic pathway of RAS, Ang II is a product of a "peptidyl dipeptidase" ACE. In this process, the decapeptide Ang I is converted by ACE to Ang II. Another carboxypeptidase, ACE2, is a key enzyme catalyzing the cleavage of both Ang I and Ang II. ACE2 cleaves the C-terminal amino acid of Ang I to the nonapeptide Ang 1~9. ACE2 also directly converts Ang II to Ang 1~7, which is act on its own receptor, Mas receptor. The conversion of Ang II to Ang 1-7 seems the preferred pathway with a 500-fold greater efficiency than that for Ang I. Emerging data implies that Ang 1~7 acts as an endogenous antagonist of the Ang II-induced actions mediated via AT1 receptors. ACE2 knockout mice showed higher blood pressure and the increased renal Ang II levels in comparison with wild-type mice31). The ACE2 knockout model also exhibited a greater degree of glomerulosclerosis and proteinuria, which was attenuated by AT1 receptor blockade32). Thus, ACE2 seems to control Ang II production by ACE, either by stimulating an alternative pathway for Ang I degradation or by promoting the convertsion of Ang II to Ang1-7 (Fig. 1).
Recent research for renin-angiotensin system and hypertension
1. Animal hypertensive model and renin-angiotensin system
Chronic infusion of low doses of Ang II provides a useful experimental model of Ang II-dependent hypertension. Ang II-infused rats have an increase in intrarenal renin, AGT, and Ang II. AT1 receptor blockade prevents these enhancements of the intrarenal RAS components in Ang II-infused hypertensive rats, indicating that the augmentation of intrarenal RAS in Ang II-dependent hypertension depends on activation of AT1 receptors33-35). Although plasma and juxtaglomerular apparatus renin are suppressed in this hypertensive model, increased distal nephron renin associated with augmented proximal tubular AGT and spillover into the distal nephron may contribute to elevated intratubular Ang I and Ang II formation as well as the progressive high blood pressure in Ang II-dependent hypertension. Moreover, the fact that AT1 receptor blockade increased plasma Ang II concentrations and decreased kidney Ang II contents by chronic Ang II infusions suggests a differential regulation of Ang II in the kidney and in the circulation35).
The importance of the intrarenal RAS in systemic hypertension has also been shown in several transgenic animal models. Lavoie et al.36) suggested that the proximal tubule RAS can have a significant effect on blood pressure using the double transgenic mouse model. They reported that dual production of renin and AGT in the renal proximal tubule can result in a systemic HTN.
In spontaneously hypertensive rats (SHR), which have been used as a model of genetic hypertension, it has been shown that augmented intrarenal AGT may contribute to increased renal Ang II and tissue damage. Although the circulating RAS generally considered to be low in SHR, several studies indicated that ACE inhibitors and/or ARBs reduces renal dysfunction, suggesting that the intrarenal RAS may be inappropriately activated and contribute to the development of HTN and HTN-induced renal injury in this animal model. Kobori et al showed that SHR have enhanced intrarenal AGT that contributes to increased Ang II levels, leading to HTN and renal injury37). AGT mRNA and protein levels in the kidney cortex and all parameters of renal damage were changed in parallel, and ARB also prevented these increases.
Because salt loading amplifies the development of HTN in strains that are predisposed to HTN, Dahl salt-sensitive rats have been used as a model of human salt-sensitive HTN. The local RAS may also be inappropriately activated and may contribute to the development of HTN in this animal model. Kobori et al showed that Dahl salt-sensitive rats fed with a high-salt diet have a paradoxical augmentation of intrarenal AGT which lead to HTN38).
A role for intrarenal Ang II, as well as beneficial effects with ACE inhibitor and ARB, has been shown in streptozotocin-induced diabetic models. In diabetic rat glomerular extracts, AGT and Ang II concentrations were increased significantly by 2.2- and 1.9-fold. Preincubation of glomerular extracts with captopril resulted in a 20~30% decrease in Ang II conversion from exogenous Ang I in diabetic and control rats39).
Activation of the intrarenal RAS has also been demonstrated in a variety of other kidney disease models and all these studies support a pathophysiologic significance of the intrarenal RAS in diverse renal injury as well as in the development and progression of HTN. Blocking the intrarenal RAS plays an important role in preventing progressive renal injury and not merely systemic HTN.
2. Clinical hypertensive studies and renin-angiotensin system
Clinical evidence also show that the intrarenal RAS markedly contributes to the development and progression of systemic HTN. Admiraal et al.40) showed intrarenal RAS activation in subjects with unilateral renal artery stenosis by tracer studies. The plasma level of de novo intrarenally produced Ang I was seven times higher on the affected kidney. They also showed intrarenal Ang II formation both in patients with essential hypertension and unilateral renal artery stenosis by estimation of Ang I-to-II conversion in the kidney. In in situ hybridization studies, there were stronger signals for renin, AGT, and ACE mRNA in mesangial and epithelial cells of kidney tissue from hypertensive patients and from patients with renal pathology characterized by mesangial proliferation. This study proposes that the RAS could be a local factor involved in the progression of chronic renal failure and consequent development of hypertension41).
The increased intrarenal generation of Ang II, despite suppression of the systemic RAS, has been demonstrated in other progressive kidney diseases. In renal mesangial cells from diabetic patients, stronger expressions for renin mRNA were observed by in situ hybridization. Immunohistochemical studies of the biopsies from 10 patients with type 2 diabetes and overt nephropathy also demonstrated increased ACE and Ang II expressions in tubular and interstitial cells42). Elevated concentrations of tubular Ang II were associated with proteinuria and interstitial cell infiltration. In patients with IgA nephropathy, glomerular expression of renin, ACE, chymase, AT1 receptors, and AT2 receptors was also increased, which correlated with the degree of mesangial hypercellularity and expansion43). Immunoreactive renin, AGT, ACE, Ang II receptor, and Ang II peptide are present in cysts and in the dilated tubules of kidneys in autosomal dominant polycystic kidney disease44). ACE-independent and chymase-mediated Ang II formation has been reported in the interstitium of kidney tissue in autosomal dominant polycystic kidney disease45). The intrarenal generation and consequent activation of RAS also plays a pathogenic role in cyclosporin A nephropathy, chronic allograft nephropathy, and in HTN of dialysis patients. Collectively, these clinical studies evidently support that activated intrarenal RAS plays a pivotal role in the pathophysiology of HTN and progressive renal diseases.
Fetal programming of hypertension - role of renin-angiotensin system
Fetal programming is the process through which adverse effects of an environmental insult early in life, particularly in utero, can predispose to adult disease. Barker et al.9) first recognized an inverse association with birth weight and death from cardiovascular disease in adulthood, indicating that some components in the perinatal environment, related in part to maternal nutrition, can "program" the individual for increased cardiovascular risk later in life. Since then, a number of studies have provided a close relationship between intrauterine events and subsequent conditions such as impaired glucose tolerance, type 2 diabetes, obesity, HTN, and chronic kidney disease.
Of these, programming of hypertension has been studied most extensively. Both epidemiological studies and experimental data have shown strong evidence that prenatal environment can modify adult blood pressure. The mechanisms linking low birth weight (LBW) and hypertension seem be multifactorial and involve alterations in the normal regulatory systems and renal functions affecting the longterm control of arterial pressure. Brenner et al.10) suggested that LBW is associated with a congenital loss in nephron number, which would result in reduced renal sodium excretion and increased susceptibility to essential HTN. This hypothesis was based on the understanding that in the setting of nephron loss, compensatory hypertrophy and hyperfiltration occurs in remaining glomeruli to sustain adequate renal function. Although the exact mechanism of the reduction in nephron number has not been clarified, possible programmed causes have been proposed; changes in DNA methylation, increased apoptosis in the developing kidney, alterations in renal RAS activity, and increased fetal glucocorticoid exposure.
It is well known that all of the components of the RAS are present in the developing kidney and play a key role in nephrogenesis. Several experimental models have shown that lower expression of the RAS components during the nephrogenic period contributes to lower nephron number and HTN in later life. Specifically, Woods et al.7) observed that maternal protein restriction during gestation in the rat was associated with a reduction in renal renin mRNA and tissue Ang II levels in the offspring at birth. Suppression of the RAS in this model was related to reduction in glomerular number, increase in arterial pressure, and decrease in GFR. Moreover, renal AT1 receptor expression was low in the newborn kidney from protein-restricted dams but rose by 28 days of age. The reduction in renal AT1 receptor at birth may play a role in mediating the reduction in nephron number and the increased expression AT1 receptor at 4 week of age suggests that upregulation of the AT1 receptor could contribute to the development of HTN8). Taken together, alterations in the intrarenal RAS may play an important role in mediating the structural alterations of kidney and HTN in later life.
Conclusion
The central role of the circulating RAS in the regulation of blood pressure and sodium homeostasis has been identified. The inappropriate activation of the intrarenal RAS and its downstream consequences is vital to many hypertensinogenic processes by a variety of pathophysiologic mechanisms. From a functional perspective, there has also been growing evidence of the organ-specific roles exerted by Ang II acting as a paracrine factor. Fetal programming of hypertension due to exposure to an adverse environment in utero may be significantly linked to alterations in the RAS.
 XML Download
XML Download