

 PDF
PDF ePub
ePub Citation
Citation Print
Print


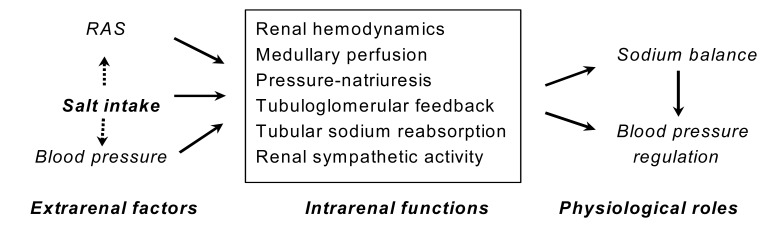
The L-arginine-nitric oxide (NO) pathway has been implicated in many physiological functions in the kidney, including regulation of glomerular hemodynamics, mediation of pressure-natriuresis, maintenance of medullary perfusion, blunting of tubuloglomerular feedback (TGF), inhibition of tubular sodium reabsorption and modulation of renal sympathetic nerve activity (Fig. 1). Its net effect in the kidney is to promote natriuresis and diuresis, contributing to adaptation to variations of dietary salt intake and maintenance of normal blood pressure.
Nitric oxide synthases
NO is produced by a reaction that is catalyzed by NO synthases (NOS). In the kidney, there have been identified all three isoforms of NOS, i.e., neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). Among them, eNOS has been described in the vascular endothelium and epithelia of certain nephron segments such as the thick ascending loop of Henle (TAL) and the collecting duct, whereas nNOS has been detected in the macula densa, efferent arterioles, Bowman's capsule, some cells of the cortical TAL, and the collecting duct. The expression of iNOS is seen in the inner medullary collecting duct possibly under basal conditions as well as in the setting of inflammation. NO does not have to be produced in a nephron segment to have an effect therein, since its high diffusibility makes it possible to affect the function of surrounding structures.
Renal hemodynamics and tubuloglomerular feedback
Studies using NOS inhibitors such as nitro-L-arginine and NG-nitro-L-arginine methyl ester (L-NAME) have shown that NO exerts a tonic influence mainly on the medullary circulation. Although the blood flow to the inner medulla comprises less than 1% of total renal flow, its changes can affect sodium and water homeostasis and long-term control of arterial pressure. An intramedullary infusion of NOS inhibitors decreases total renal blood flow, renal interstitial fluid pressure, and urine volume and sodium excretion, without significantly altering glomerular filtration rate, fractional sodium and water excretion, blood pressure, or urine osmolality. Furthermore, an intravenous infusion of angiotensin II (AII), norepinephrine or vasopressin at doses that are normally subpressive causes hypertension in the presence of intramedullary infusion of L-NAME.
On the other hand, NO is an important modulator of TGF responsiveness, if not a direct mediator. NO synthesized in the macula densa by the action of nNOS attenuates TGF-mediated constriction of afferent arterioles. Conversely, a blockade of nNOS may sensitize TGF responsiveness, leading to renal vasoconstriction, sodium retention and arterial hypertension. An exaggerated TGF response is diminished by angiotensin II type 1 receptor (AT1R) blockade or by treatment with tempol. An enhancement of NO production may also be responsible for the impairment of autoregulatory efficiency in medullary blood flow during volume expansion.
Dietary salt intake
Renal NO synthesis plays a role in acute and chronic regulation of sodium balance. In rats maintained on high salt diet, the expression of all NOS isoforms is increased in the inner medulla, while not significantly altered in the outer medulla and cortex. However, the most consistent finding in response to an altered dietary salt intake is a change of nNOS expression in the macula densa: a dietary salt restriction increases the expression of nNOS in the cortex and macula densa, whereas a high salt diet reduces macula densa nNOS expression. Blockade of nNOS with 8-nitroindazole prevents the effects of salt intake on nitrite excretion or the renal vascular responses to L-NAME.
Overall, the dietary salt loading increases the plasma concentration and the urinary excretion of nitrites. The endogenous NO participates in the renal adaptation to increased dietary salt intake, facilitating sodium excretion and allowing maintenance of normal blood pressure. Among others, the mechanisms underlying the changes of dietary salt intake altering the renal expression of NOS may involve the activity of endothelin (ET) system. A high salt diet may increase outer medullary osmolality, which in turn increases ET-1 release and stimulates eNOS expression in TAL through activation of endothelin B (ETB) receptors.
Tubular sodium transport
NO has an inhibitory effect on tubular sodium reabsorption, resulting in enhanced urinary excretion of water and solutes. The effect of NO on tubular sodium transport may vary in different segments of the nephron. The inhibitory effect of NO on proximal tubular sodium reabsorption may be attributed to decreased apical Na+/H+ exchange and reduced Na+,K+-ATPase activity. In TAL, NO reduces sodium reabsorption by inhibiting Na+/K+/2Cl- cotransporter (NKCC2) and inhibits bicarbonate reabsorption by reducing Na+/H+ exchange activity. eNOS is the isoform responsible for NO synthesis leading to the inhibition of NaCl reabsorption in TAL. NKCC2 is also expressed on the luminal surface of the macula densa and appears to be responsible for monitoring distal delivery, which is detected as a change in chloride concentrations.
The tubular sodium reabsorption may also be affected by way of amiloride-sensitive transport process, suggesting that the distal tubule is an important site of NO action. NO formed in collecting duct cells also directly affects tubular sodium transport by inhibiting amiloride-sensitive epithelial sodium channels. In addition, NO inhibits vasopressin-stimulated osmotic water permeability in the cortical collecting duct by a mechanism involving guanylyl cyclase-dependent increase of cyclic guanosine 3',5'-monophosphate (cGMP) which leads to a decrease of cytosolic cyclic adenosine 3',5'-monophosphate (cAMP). It also inhibits H+-ATPase activity in acid-secreting intercalated cells of the cortical collecting duct and reduces urea transport in the inner medullary collecting duct.
Pressure-natriuresis
NO has been implicated in mediating the sodium excretion that occurs in response to increases of renal arterial pressure. The increased perfusion pressure stimulates NO synthesis due to increased endothelial cell shear stress, and the urinary amount of nitrite excretion positively correlates with the sodium excretion. While an enhancement of renal tissue NO levels may directly inhibit distal tubular sodium transport, a reduction of NO activity may cause right-shift of the pressure-natriuresis curve to result in hypertension.
Interactions with sympathetic nerves
The sympathetic nervous system plays a role in the regulation of renal hemodynamics and sodium homeostasis. Basal NO synthesis may blunt the sympathetic vasoconstrictive effect in the isolated perfused rat kidney. The relative insensitivity of the medullary circulation to the renal sympathetic activation is attributed to the medullary NO synthesis. NO also mediates the increased proximal tubular reabsorption of sodium that occurs with renal sympathetic nerve stimulation, which is in fact a rare example where NO is actually increasing the sodium reabsorption.
Interactions with O2- and angiotensin II
An increase in reactive oxygen species (ROS) activity causes upregulation of NOS expression in human coronary artery endothelial cells, which appears to be in part mediated by limiting the availability of NO and thereby exerting a negative feedback influence on NOS expression. A balance between NO and O2- contributes to the maintenance of normal kidney function by opposing each other's action: an enhanced production of O2- can influence kidney function either directly or indirectly via a reduction in NO bioavailability, leading to sodium retention. NO itself also acts as a powerful anti-oxidative agent that minimizes the adverse effects of O2. In addition, NO donor diethylenetriamine-NO (DETA-NO) has been found to increase the expression of extracellular superoxide dismutase (ecSOD) in human aortic smooth muscle cells, which may represent an important feed-forward mechanism, whereby endothelial NO stimulates ecSOD expression in adjacent smooth muscle cells to prevent O2--mediated degradation of NO as it traverses between the two cell types.
Inhibition of NOS not only decreases basal luminal diameters of arterioles but also augments constrictor responses to AII, suggesting that NO antagonizes the constrictor action of AII. AII may activate the renal production of bradykinin and NO via stimulation of angiotensin II type 2 receptor (AT2R). AII also induces NO release from nNOS via AT1R in macula densa cells in rabbits. While NO from eNOS regulates basal blood flow in the cortex and medulla, NO produced by nNOS mediates the increase of medullary blood flow in response to AII. A recent study also showed that NO derived from eNOS mediated AII-mediated facilitation of norepinephrine release in response to renal nerve stimulation. NO bioavailability may also be influenced by AII-induced activation of ROS which quench the released NO. An acute administration of AII in dogs markedly reduces glomerular filtration rate after NOS inhibition, but not in the intact NO condition, which was attenuated by concomitant administration of O2- scavenger tempol.
The greater sensitivity of efferent arterioles to AII is related to a weaker influence of NO on these vessels. Another important area of interaction between NO and AII is in the control of TGF sensitivity. AII is a powerful stimulus to sensitize TGF, whereas NO has an opposing effect on TGF sensitivity. An overweight of AII over NO will lead to vasoconstriction and body fluid volume retention, resulting in an increase of arterial pressure. Conversely, a dominance of NO will strongly desensitize TGF, giving vasodilation, increased fluid volume excretion and reduction of blood pressure.
Pathophysiological implications of nitric oxide
1. Chronic kidney diseases
Chronic renal disease is characterized by a deficiency of NO bioactivity. The plasma from uremic patients has been shown to inhibit L-arginine transport in cultured endothelial cells1). In rats with 75% renal mass reduction and saline load for 4 weeks, NO excretion and NOS activity decreased along with the hypertension2). Moreover, eNOS mRNA, eNOS protein and eNOS activity are all decreased in human endothelial cell cultures exposed to erythrocytes from patients with end-stage renal disease3). The plasma level of asymmetric dimethylarginine (ADMA, endogenous inhibitor of NOS) is positively correlated with systolic blood pressure in 5/6 nephrectomized rat model4), in which the increase of ADMA may be attributed to upregulation of protein methyltransferase, which catalyzes formation of ADMA, and downregulation of dimethylarginine dimethylaminohydrolase, which metabolizes ADMA.
2. Acute kidney injury
Benefits of providing NO donors before (but not following) the ischemic injury have been well documented. A pretreatment with NO donor molsidomine or L-arginine ameliorated the renal injury in ischemic acute renal failure5). Provision of L-arginine at the time of ischemic injury also attenuated the reduction of glomerular filtration rate. Similarly, pretreatment with NO donor FK409 was shown to mitigate the ischemic renal injury, whereas treatment after ischemia worsened it6).
3. Hypertension
1) Spontaneous hypertension
In spontaneously hypertensive rats (SHR), the renal expression of NOS and the urinary excretion of NO are increased at 9-12 weeks of age7). The hypertension could be exaggerated by NOS inhibition. An enhanced O2- production and its interaction with NO play an important role in the pathogenesis of salt sensitivity in SHR. A two-fold increase of renal cortical nitrotyrosine deposition suggests an enhanced interaction of NO with ROS in SHR8), in which AII is implicated in increases of ROS and decreases of SOD. In SHR, the activity of antioxidant enzymes is diminished, and anti-oxidant treatment increases NO bioavailability to reduce the arterial pressure.
Nevertheless, the regulation of NO differs according to the age of the animal. With advanced age, SHR show reduced urinary NO excretion and depressed expression of NOS proteins in the kidney9). The decreased expression of NOS underscores the importance of these enzymes in the pathophysiology and maintenance of hypertension.
2) Dahl salt-sensitive hypertension
A reduced synthesis of NO in the renal medulla has been implicated in the pathogenesis of Dahl salt-sensitive (DS) hypertension. In response to salt loading, DS rats cannot but Dahl salt-resistant (DR) rats may markedly increase renal NOS activity10). In the kidney of DS rats, the activity of nNOS is decreased with its mRNA expression reduced11).
L-Arginine prevented the development of hypertension and normalized the pressure-natriuresis relationship in DS rats12). A selective iNOS inhibition with aminoguanidine augmented the increase of arterial pressure in DS rats on high salt intake13). Conversely, compensatory upregulation of iNOS expression and NOS activity may represent a novel mechanism regulating the increased volume load and blood pressure in DR rats. However, the activity eNOS in the kidney of DS rats seems to be unaltered.
Furthermore, DS rats on high-salt diet showed a significant correlation between the urinary excretion of ADMA and the degree of hypertension, suggesting that ADMA play a role in the pathogenesis of hypertension14). The hypertension in DS rats is also associated with renal artery hypertrophy, increased vascular and renal ET-1 protein contents, and glomerulosclerosis10).
The administration of tempol attenuates DS hypertension, indicating that the hypertension is also in part related to an increase in oxidative stress and a decrease in anti-oxidant capacity. It is indeed associated with marked decreases in medullary SOD activity and increases in oxidative tissue damage15).
3) Deoxycorticosterone acetate-salt hypertension
Deoxycorticosterone acetate (DOCA)-salt treatment increases the urinary excretion of nitrite and nitrate, along with the elevation of blood pressure16). NOS inhibition exaggerates the hypertensive response, whereas L-arginine markedly reduces the blood pressure and improves the cardiovascular dysfunction in DOCA-salt rats17). However, there may be uncoupling of NOS/NO and soluble guanylyl cyclase (sGC)/cGMP pathways in the inner medulla in this model of hypertension18). Although the urinary cGMP excretion increases after 1 week of DOCA-salt treatment, it gradually decreases to restore the normal values by the third week16). An attenuation of the hypertensive response by anti-oxidant treatment was also noted in DOCA-salt rats19).
4) NG-nitro-L-arginine methyl ester hypertension
An infusion of NG-nitro-L-arginine methyl ester (L-NAME) into the renal medulla of rats causes sodium retention and arterial hypertension. A strong negative correlation exists between NOS activity and blood pressure in rats chronically treated with L-NAME. Its hemodynamic changes have been primarily attributed to an abrupt withdrawal of tonic vasodilator effect, leaving unopposed of an equally tonic action of endogenous vasoconstrictors. However, the development of hypertension represents a complex mechanism which may not simply be attributed to an inactivation of NO system. The hypertension could be partly reversed by inhibition of vasoconstrictors such as AII, vasopressin or ET20). The circulating plasma renin activity may bear no consistent relationship with the protective action of chronic AII blockade, because the plasma renin activity was increased, unchanged or decreased in this model of hypertension.
In addition, NOS inhibition increases O2- activity and increases the urinary excretion of 8-isoprostane (a marker of enhanced O2- activity)21). The exaggerated hypertensive response during high-salt intake is abolished by concomitant administration of tempol19). It is suggested that O2- contributes to the development of salt sensitivity in L-NAME hypertension.
Liu et al.22) observed no changes of plasma vasopressin levels in animals chronically treated with L-NAME. However, although administration of L-arginine and NO donors have variable effects on vasopressin secretion, the most common one is inhibition, and blockade of NO synthesis increases vasopressin secretion23). Furthermore, the hypertension is attenuated by inhibition of vasopressin20). In this context, it is worth to note an upregulation of aquaporin-2 (AQP2) in the kidney in L-NAME hypertension24), which may in fact be related with altered vasopressin levels. On the contrary, a more recent study demonstrated a downregulation of AQP2, which may represent compensatory effects increasing the urinary excretion25).
An increased sympathetic drive may also be involved in the development of L-NAME hypertension. Acute ganglionic blockade produced a large fall of blood pressure, and chronic sympathectomy by daily injections of ganglionic blockers attenuated the hypertension. Furthermore, chronic bilateral renal denervation delayed and attenuated the hypertension26). In this context, water channels may also be affected in L-NAME-treated rats, since some water channels are under tonic excitatory influence of the sympathetic nerves27). On the contrary, it has been found that the hemodynamic actions of acute NOS inhibition in the unstressed rat are independent of the sympathetic nervous system. A more recent study also failed to demonstrate changes in renal sympathetic nerve activity in L-NAME hypertension in conscious rabbits, indicating that the renal nerves do not mediate the increase of blood pressure28).
Prostacyclin (PGI2) inhibits NO release from cultured endothelial cells29). In the dog kidney, the full renal vasodilator potential of NO (or PGI2) is expressed only in the presence of prostaglandin (or NO) inhibition, suggesting that these autacoids are mutually antagonistic. It is thus likely that some of the constrictor responses seen with chronic NOS inhibition may result from enhancement of the actions of vasoconstrictor arachidonic acid products. Nevertheless, acute cyclooxygenase inhibition does not affect the hypertension in rats treated with L-NAME, although additional renal vasoconstriction does occur30).
5) Two-kidney, one-clip hypertension
Chronic two-kidney, one-clip (2K1C) hypertension has been associated with an impaired endothelium-dependent vasorelaxation31). Both early and chronic phases of 2K1C hypertension are augmented by NO synthesis inhibitors. Furthermore, 2K1C hypertension is associated with reduced levels of eNOS proteins in the medulla of both clipped and contralateral kidneys, being causally related with reduction of medullary blood flow32). In the early reversal of hypertension and changes of renal function following unclipping of the clipped artery, an upregulation of eNOS and enhanced release of NO is an important component33). It is likely that NO contributes a significant vasodilator tone to buffer the hypertension and maintains perfusion of kidneys in this model.
6) Angiotensin II-induced hypertension
NO has been shown to provide renoprotection in AII-induced hypertension. Salt-sensitive hypertension develops after short-term exposure to AII, in association with a loss of intrarenal NO formation that could alter the ability of the kidney to excrete a salt load34). The sodium-retaining effect of AII in the absence of NO is mediated mainly by enhanced O2- activity35), suggesting that an interaction between NO and O2- in the kidney plays an important role in the development of salt sensitivity and hypertension. In fact, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase - one important source for free superoxide - was activated and the superoxide production was enhanced due to AII10). AII can also reduce ecSOD expression36).
In rats with 2-kidney, 1 figure-8 wrap (Grollman) hypertension, AII mediates renal production of bradykinin, which, in turn, releases NO and cGMP via stimulation of AT2R37). Aortic banding resulted in significant upregulation of eNOS, iNOS and nNOS in the renal cortex and medulla38). The aortic coarctation-induced hypertension is associated with increased nitrotyrosine abundance in all tissues exposed to high arterial pressure, denoting enhanced ROS-mediated inactivation and sequestration of NO39).
7) Other models of hypertension
Table 1 summarizes the alterations of NO system in the kidney in various models of hypertension. A chronic consumption of a high salt diet results in moderate hypertension, which is accompanied by downregulation of various NOS isoforms that undoubtedly contributes to the development and maintenance of hypertension40). In fructose-fed rats, while the vascular expression of NOS proteins is not altered41), the medullary NO production is reduced during high sodium load, resulting in salt-sensitive hypertension42). The renal expression of iNOS and eNOS mRNA is reduced in hypertension induced by adrenocorticotropic hormone (ACTH) or corticosterone43). A chronic exposure to low levels of lead results in a marked elevation of blood pressure, in association with significant reduction in urinary NO excretion and upregulation of eNOS and iNOS abundance in the kidney44). Chronic elevation of plasma vasopressin levels enhances the medullary eNOS protein expression, which enables sustained elevations of NO concentrations to buffer the hypertensive effects of vasopressin45). It has been also found that the hypertensive effect of intravenous infusion of norepinephrine was chronically buffered by the α2 receptor-mediated increase of NOS activity within the renal medulla46). In hydronephrotic rats, a reduced NO availability through reduced expression of nNOS and eNOS proteins in the diseased kidney and subsequent resetting of TGF mechanism plays an important role in the development of hypertension47). Although eNOS protein and tissue nitrate contents are decreased along with enhanced ET-1 proteins in the aorta of type II 11β-hydroxysteroid dehydrogenase-deficient rats48), their changes in the kidney have not been documented. Oral contraceptive users exhibit elevated AII levels and AT1R expression, of which hemodynamic effects may be modulated by an increased activity of NO pathway49).

 XML Download
XML Download