

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
In the kidney, prostaglandins play a role in regulating renal hemodynamics, renin release, and tubular sodium and water reabsorption1-3). The main renal prostaglandin (PG) is prostaglandin E2 (PGE2) which is synthesized from arachidonic acid in the presence of cyclooxygenase isoforms cyclooxygenase-1 (COX-1) or cyclooxygenase-2 (COX-2) with the prostaglandin E synthase (PGES)4). The effects of PGE2 on renal water handling have widely been investigated5). Previous studies revealed that PGE2 plays a role in modulating the effect of vasopressin on osmotic water permeability in the renal collecting duct where it attenuates the anti-diuretic action of vasopressin. For example, 1) PGES inhibits both cAMP synthesis and elevation of cytosolic Ca2+ in rabbit cortical collecting ducts5) and rat terminal inner medullary collecting ducts, causing decreased trafficking of aquaporin 2 (AQP2 to the apical plasma membrane6, 7); 2) PGE2 stimulation induces endocytosis of AQP2 in forskolin-stimulated Madin-Darby canine kidney (MDCK) cells, suggesting that PGE2 induces internalization of AQP2 independently of AQP2 dephosphorylation6); and 3) the diuretic effect of PGE2 also includes the cAMP and Ca2+-independent activation of the Rho-kinase and formation of F-actin8).
Cyclooxygenase is the major rate-limiting enzyme in the cascade leading to the synthesis of PG from arachidonic acid. COX-1 and COX-2 have similar catalytic activity. However their amino acid sequence and localization in the kidney are different1, 4). COX-1 is believed to be constitutively expressed, whereas COX-2 is considered only to be induced during physiological stress1, 9). Consistent with this, COX-2 expression has been demonstrated to be regulated by the interstitial osmolality in the inner medulla both in vivo and in vitro9). Moreover, COX-2 expression is upregulated by dehydration in both normal rats1). However, renal localization and regulation of these enzymes in conditions with water and sodium balance disturbance (e.g., diabetes insipidus) are not clearly understood. Lithium is an important drug widely used in the management of bipolar affective disorders10). Lithium treatment, however, is associated with polyuria and decreased urinary concentrating ability in patients10). The impaired urinary concentration ability and polyuria have been associated with impaired V2-receptor-mediated vasopressin responsiveness11) and decreased protein abundances of AQP2 and aquaporin 3 in collecting duct principal cells12). Moreover, LiCl administration in rats induced a significant excretion of urinary PGE2 along with polyuria and nonselective cyclooxygenase inhibitor, indomethacin treatment to rats with lithium-induced nephrogenic diabetes insipidus (Li-NDI) diminished urine volume and urinary PGE2 levels13), suggesting that cyclooxygenase-induced PG could be, at least in part, involved in the pathogenesis of Li-NDI.
The purposes of the present study were therefore to examine 1) the changes of expression and cellular localization of COX-2, the major PGE2 synthesis enzyme, in the kidney inner medulla and cortex + outer medulla in rats with Li-NDI (vs. control rats); and 2) the changes of urinary excretion of PGE2 in rats with Li-NDI (vs. control rats).
Materials and Methods
1. Experimental animals
The studies were performed on adult male Sprague-Dawley rats initially weighing 180-200 g (Hyochang Science, Daegu, Korea). The rats were maintained on a standard rodent diet with free access to water before the experiments.
2. Experimental protocols
Lithium chloride was added to the chow to give a concentration of 40 or 60 mEq lithium/kg of dry food as previously described12, 14). Rats received food containing 40 mEq lithium/kg of dry food for the first 7 days and thereafter, 60 mEq lithium/kg of dry food for the following 3 weeks. Previously, we demonstrated that this protocol resulted in plasma lithium levels at therapeutic levels (0.8-1.3 mM) and minimized the weight loss caused by lithium14, 15). For the last 7 days, both lithium-treated rats (n=6) and control rats (n=6) were maintained in metabolic cages to allow daily measurement of urine output and sample collections. They were supplied with the same amount of food per day (lithium-treated rats: 7 g/100 g of body weight of food containing 60 mEq lithium/kg of dry food or control rats: 7 g/100 g of body weight of food without lithium) to give a constant daily sodium intake. The rats were fed once daily in the morning, and ate nearly all of the offered food during the course of the day. All rats had free access to water intake.
3. Semiquantitative immunoblotting
All rats were killed under light enflurane anesthesia and the right kidneys were rapidly removed, dissected into two parts (i.e., cortex + outer medulla combined and inner medulla) and homogenized in ice-cooled isolation solution (0.3 M sucrose, 25 mM imidazole, 1 mM EDTA, 8.5 uM leupeptin, 1 mM phenylmethylsulfonyl fluoride, pH 7.2) using an Ultra-Turrax T8 homogenizer (IKA Labortechnik, Staufen, Germany). To remove whole cells, nuclei and mitochondria, the homogenates were centrifuged at 4,000 g for 15 minutes at 4℃ and the supernatant was pipetted off and kept on ice for further processing. The total protein concentration was measured (Pierce BCA protein assay reagent kit, Pierce, Rockford, IL) and all samples were adjusted with isolation solution to reach identical final protein concentrations. They were solubilized at 65℃ for 15 minutes in Laemmli sample buffer, and then stored at -20℃. To confirm equal loading of protein, an initial gel was stained with Coomassie Blue dye, as described previously12, 14). SDS-PAGE was performed on 12% polyacrylamide gels. The proteins were transferred, incubated with antibodies, and the sites of antibody-antigen reaction were visualized, as previously described12, 14). The band densities were quantitated by scanning the films and the density was calculated as a fraction of the mean control value for that gel.
5. Primary antibodies
For both semiquantitative immunoblotting and immunohistochemistry, previously well characterized anti-COX-2 polyclonal antibody (Cayman Chemical, Ann Harbor, MI) was used. Anti-rat AQP2 antibody (7661AP) was kindly presented by Dr. S. Nielsen, University of Aarhus, Denmark.
6. Measurements of urinary PGE2
An enzyme immunoassay kit (Amersham Pharmacia Biotech, Little Chalfont, UK) was used to determine PGE2 in the urine of experimental animals. The assay was preformed according to manufacturer's instructions. Fifty µL of urine sample or standard solution was mixed with assay buffer (0.1 M phosphate buffer pH 7.5, 0.9% bovine serum, 0.5% Kathon), PGE2 antiserum and PGE2-horseradish conjugate in provided microplate. The mixture was incubated at room temperature for 1 h on microplate shaker. After washing the enzyme, substrate (3,3',5,5' tetramethylbenzine/hydrogen peroxide) was applied to the microplate for 30 minutes. After stopping the reaction with 1 M sulphuric acid, optical density was measured at 450 nm. A standard curve was obtained using optical densities of standard samples and was used to determine the PGE2 concentrations in the urinary samples.
Results
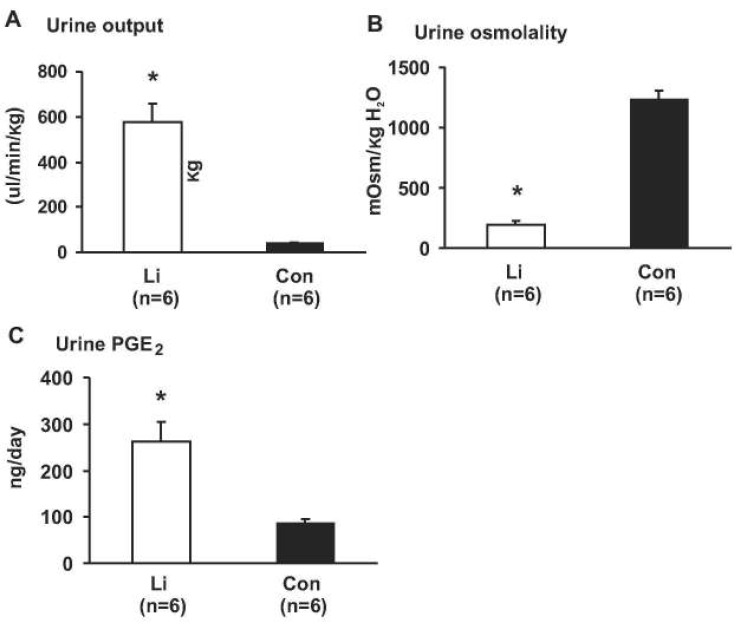
1. Chronic lithium treatment in rats was associated with polyuria, significantly decreased urine concentration and increased urinary PGE2 excretion
Chronic LiCl treatment of rats for 4 weeks was associated with markedly increased urine output, compared with controls (578±83 vs. 43±3 µL/min/kg, p<0.05, Fig. 1A). Consistent with this, urine osmolality was significantly decreased in rats with Li-NDI (190±30 vs. 1,239±70 mOsm/kg H2O, p<0.05, Fig. 1B), indicating that chronic lithium treatment in rats resulted in an impaired urinary concentration, as previously demonstrated12, 14, 15). The 24-hour urinary PGE2 excretion was significantly increased in rats with Li-NDI compared with control rats (262±42 vs. 88±7 ng/day, p<0.05, Fig. 1C).
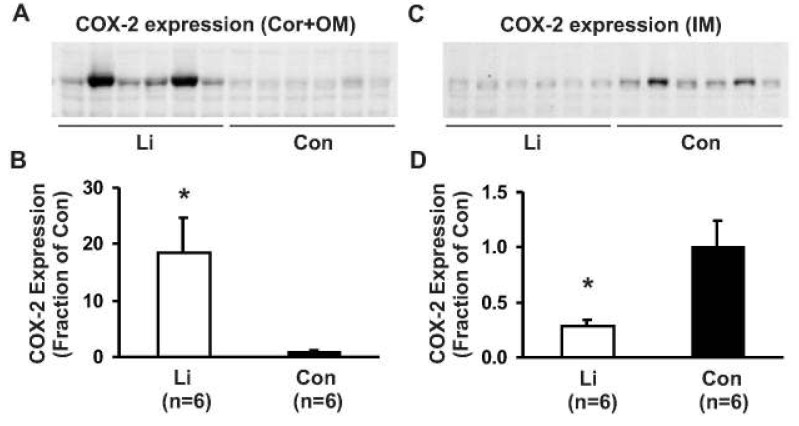
2. Chronic lithium treatment in rats was associated with increased COX-2 protein expression in the kidney cortex and decreased COX-2 expression in the inner medulla
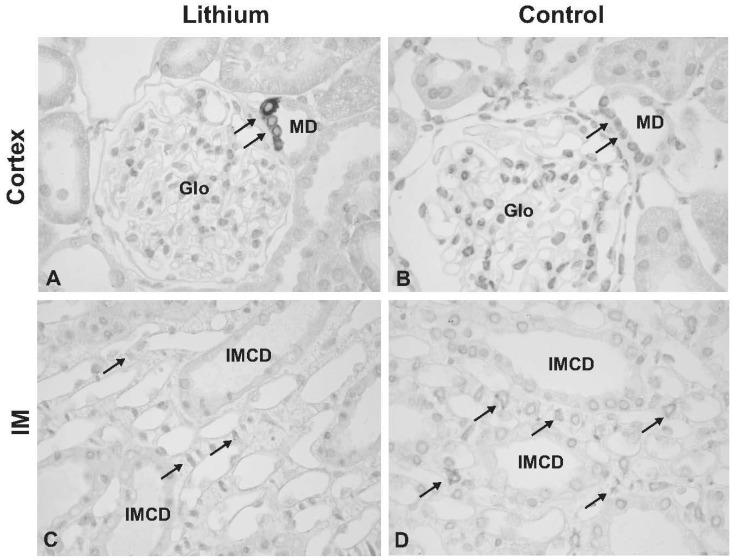
Chronic LiCl treatment markedly increased the expression of COX-2 in the kidney cortex and outer medulla to 1,845±618% of the control levels (100±18%, p<0.05, Fig. 2A, B). In contrast, COX-2 expression was significantly decreased in the kidney inner medulla to 28±7% of the control levels (100±24%, p<0.05, Fig. 2C, D). Immunoperoxidase lebeling of COX-2 demonstrated an increased COX-2 labeling in the macula densa region of the cortical thick ascending limb in lithium-treated rats (indicated as MD in Fig. 3A) compared with the controls (Fig. 3B). Moreover, the number of COX-2 positive tubular cells was increased in lithium-treated rats (not shown). In the outer medulla, COX-2 immunolabeling was not observed in both lithium-treated rats and control rats (not shown), consistent with previous studies17). In the inner medulla, weak but distinct COX-2 labeling was observed in a characteristic perinuclear pattern in inner medullary interstitial cells of kidneys from control rats (arrows in Fig. 3D), whereas no or very weak labeling of COX-2 was seen in the inner medulla of kidneys from lithium-treated rats (arrows in Fig. 3C). In contrast, COX-2 labeling was not seen in the collecting duct cells in both lithium-treated rats and control rats (Fig. 3C, D).
Discussion
The present study demonstrated that 1) there was a differential regulation of COX-2 between cortex and inner medulla in Li-NDI, thus, Li-NDI was associated with significantly decreased expression of COX-2 in the inner medullary interstitial cells, whereas COX-2 expression at the macula densa in the cortical thick ascending limb was markedly increased; and 2) urinary PGE2 excretion was significantly increased in Li-NDI, possibly due to an increased renal production of PGE2.
We demonstrated altered expression of renal COX-2 in rats with Li-NDI where urine concentration was significantly decreased. Li-NDI could be associated with reduced interstitial osmolality, as previously evidenced by the decreased expression of urea transporters UT-A1 and UT-B, and vasopressin-stimulated phosphorylation of UT-A118). Decreased inner medullary COX-2 expression observed in the present study is consistent with previous studies raising the possibility that decreased interstitial osmolality in inner medulla may play a role in the downregulation of inner medullary COX-2 expression9). Moreover, recent studies demonstrated that dehydration resulted in an increase of inner medullary COX-2 expression in both Sprague-Dawley rats9) and in rats with Li-NDI17), paralleled by an increased urine concentration. Therefore, the findings of decreased expression of inner medullary COX-2 in polyuric Li-NDI (present study) and the increased expression of COX-2 in response to dehydration9, 17) may suggest that COX-2-derived prostaglandins could, at least in part, participate in the regulation of urinary water excretion.
In contrast to the down-regulation of COX-2 in the inner medulla, COX-2 expression in the kidney cortex was markedly increased, and the number of COX-2-labeled cells in the macula densa region of the ascending thick ascending limbs was increased by lithium treatment. In the macula densa region, it has been demonstrated that an increased COX-2 expression is associated with conditions exhibiting high renin levels, e.g., sodium restriction, angiotensin converting enzyme inhibition, and renovascular hypertension1). In contrast, selective COX-2 inhibitors significantly decrease plasma renin levels and renal renin activity1). Thus, the regulation of renin secretion in the juxtaglomerular apparatus mediated by macula densa could be dependent on the COX-2-derived prostanoids. Previous studies demonstrated that rats with Li-NDI are associated with an increase in urinary sodium excretion mainly due to dysregulation of epithelial sodium channel ENaC subunits in addition to the polyuria and impaired urinary concentration14), thereby they are prone to the development of extracellular fluid volume contraction and hence high renin secretion. The increased level of COX-2 expression and the increased number of COX-2-labeled cells in the macula densa region are therefore consistent with the state of potential extracellular fluid volume contraction in Li-NDI.
We demonstrated that 24-hour urinary excretion of PGE2 was significantly increased in rats with Li-NDI when compared with control rats. This is consistent with previous findings showing an increased urinary PGE2 excretion in Li-NDI in both experimental animals and human patients as well as in patients with congenital forms of NDI13, 19). Since COX-2 expression in the kidney cortex was increased while the expression was decreased in inner medulla, it could be possible that measurements of PGE2 in the urine do not reflect the inner medullary renal PGE2 synthesis accurately or cortical COX-2 expression and PGE2 production may mainly affect the urinary PGE2 excretion. Thus, further studies are needed to understand the discrepancy between the increased urinary PGE2 excretion and decreased expression of inner medullary COX-2 in Li-NDI.
The mechanisms by which lithium induces diabetes insipidus are incompeletely understood. This study demonstrated that urinary PGE2 excretion is increased, suggesting that Li-NDI could be associated with an increased renal production of PGE2. There is a number of evidence that PGE2 directly inhibits solute reabsorption in microperfused thick ascending limbs as well as water and solute reabsorption in the collecting ducts20-22). Moreover, acute infusion of PGE2 into the normal animals has been shown to cause water diuresis 3 and to inhibit water absorption in collecting ducts5). PGE2 therefore appears to antagonize the hydrosmotic effect of vasopressin and thus PGE2 may counteract vasopressin action in Li-NDI, serving as local feedback regulators of the anti-diuretic action of vasopressin. Consistent with this, a recent study demonstrated that COX-2 inhibition completely prevented lithium-induced polyuria and an increased urinary PGE2 excretion in COX-1 null mice (COX-1 -/- mice), suggesting that COX-2 and/or COX-1-induced increase in the PGE2 production could play a role in the lithium-induced polyuria23).
In summary, 1) there is a differential regulation of COX-2 between cortex and IM in Li-NDI; and 2) urinary PGE2 excretion is markedly increased in Li-NDI, possibly due to an increased renal production. PGE2 is therefore likely to play a role in modulating the effect of vasopressin on the water reabsorption in the renal collecting duct where it attenuates the anti-diuretic action of vasopressin in Li-NDI.
 XML Download
XML Download