

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Potassium is the most abundant cation in the body. Although extracellular potassium accounts for only 2% of total body potassium, it has a major effect on the ratio of intracellular to extracellular potassium and on the resting membrane potential. As a result, serum potassium is normally regulated around the narrow range of 3.5-5.0 mEq/L.
Hypokalemia is a common clinical problem. Decreased intake, increased translocation into the cells, or increased losses in the urine (or gastrointestinal tract or sweat) all can lead to hypokalemia. The kidney is the major route of potassium excretion, accounting for 90% of potassium loss daily. The remaining 10% is excreted through the gastrointestinal tract. The kidney is therefore responsible for long-term potassium homoeostasis, as well as the serum potassium concentration.
The main nephron site where K secretion is regulated is the cortical collecting duct (CCD), mainly via the effects of aldosterone. Transport studies in this tubule segment have demonstrated that potassium secretion is accomplished via apical potassium channels (ROMK: luminal, ATP-regulated, inwardly rectifying K channel). The secretion of potassium in this nephron segment is indirectly but tightly coupled to sodium reabsorption via the amiloride-sensitive sodium channel; increased sodium reabsorption increases whereas decreased sodium reabsorption decreases potassium secretion. Aldosterone interacts with the mineralocorticoid receptor (MR) to increase sodium reabsorption and potassium secretion; the removal of cationic sodium makes the lumen relatively electronegative, thereby promoting passive potassium secretion from the tubular cell into the lumen through apical potassium channels (Fig. 1). As a result, any condition that decreases the activity of renal potassium channels results in hyperkalemia (for example, amiloride intake or aldosterone deficiency) whereas their increased activity results in hypokalemia (for example, primary aldosteronism or Liddle's syndrome)1, 2). Increased aldosterone, in addition, can cause metabolic alkalosis by increasing bicarbonate reabsorption in the collecting duct.
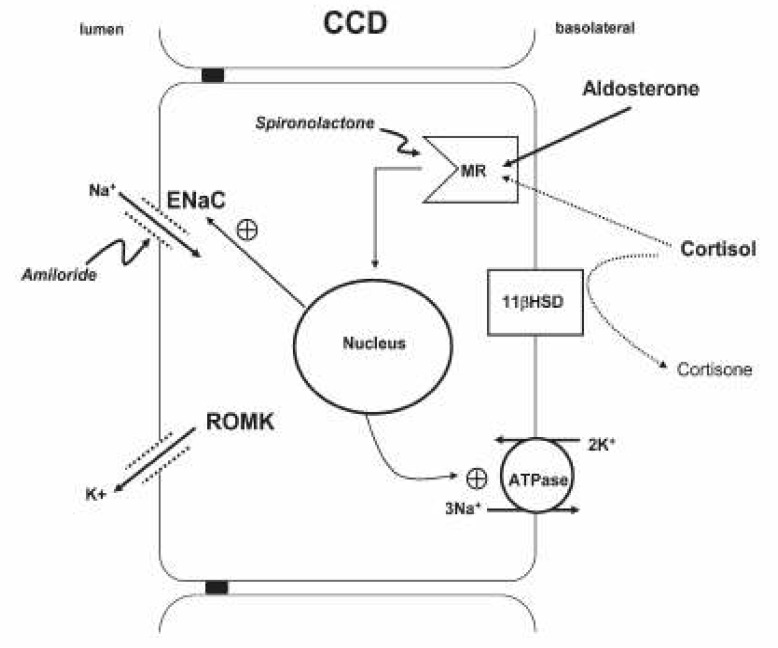 | Fig. 1K+ transport at the principal cell of the cortical collecting duct (CCD). MR, mineralocorticoid receptor; 11β-HSD, 11β-hydroxysteroid dehydrogenase; ENaC, epithelial Na channel; ROMK, luminal ATP-regulated inwardly rectifying K channel. [From Landau D : Cell Mol Life Sci 63:1962-1968, 2006]
|
The cause of hypokalemia can usually be determined from the history. If there is no apparent cause, the initial step is to see if hypokalaemia is in association with systemic hypertension or not. In the former group hypokalaemia is associated with a high mineralocorticoid effect or hyperactive sodium channel as in Liddle's syndrome. In normotensive patients, hypokalaemia could be secondary to overt or occult gastrointestinal loss or due to renal potassium wasting (Table 1). In hypertensive hypokalemic patients, measurement of the renin, aldosterone, and cortisol concentrations would be of help in differential diagnosis.
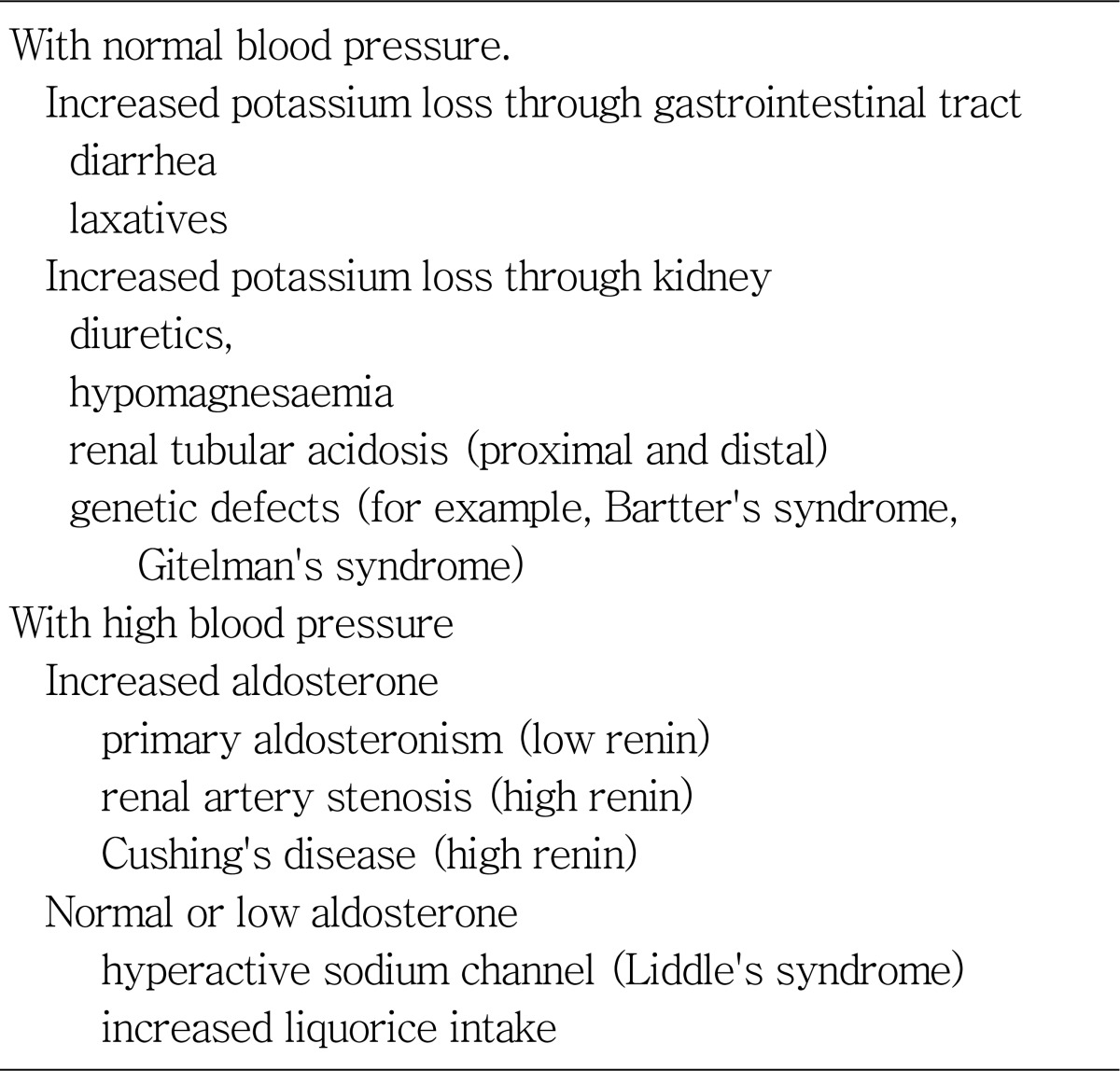
Table 1
Work-up of Chronic Hypokalemia (more than 24 hours of onset)
[Modified from Rastergar A, Soleimani M : Postgrad Med J 77:759-764, 2001]
![]()
This review discusses the biology of aldosterone and cortisol followed by a discussion of several forms of hypertensive hypokalemic disorder.
Go to : 
Aldosterone and cortisol synthesis
Aldosterone is synthesized in the zona glomerulosa of the adrenal cortex. In the zona glomerulosa progesterone is hydroxylated at C21 by CYP21A2 to yield deoxycorticosterone. The three final steps in the conversion of this intermediate to aldosterone (11-beta-hydroxylation, 18-hydroxylation, and 18-methyl oxidation) are catalyzed by a single mitochondrial P450 enzyme (aldosterone synthase, CYP11B2, P450c11as) encoded by the gene CYP11B2. A similar, highly homologous enzyme, CYP11B1 (encoded by the gene CYP11B1), acts in parallel in the zona fasciculata to convert 11-deoxycortisol to cortisol. The absence of aldosterone synthase in the zona fasciculata prevents aldosterone secretion from being regulated by ACTH. The human CYP11B2 is encoded by a gene closely related to CYP11B1, with over 90 percent homology at the protein level. The CYP11B2 gene is located on chromosome 8q24.3.
The last step in cortisol biosynthesis is the 11-beta-hydroxylation of 11-deoxycortisol, a reaction catalyzed by the mitochondrial enzyme CYP11B1. CYP11B1 is also encoded by a gene on chromosome 8q24.3, near the gene CYP11B2. The product of the CYP11B1 gene is a 51 kDa protein that is nearly identical to the CYP11B2 enzyme responsible for the terminal steps of aldosterone synthesis, except that it lacks the ability to convert corticosterone to aldosterone (Fig. 2)3).
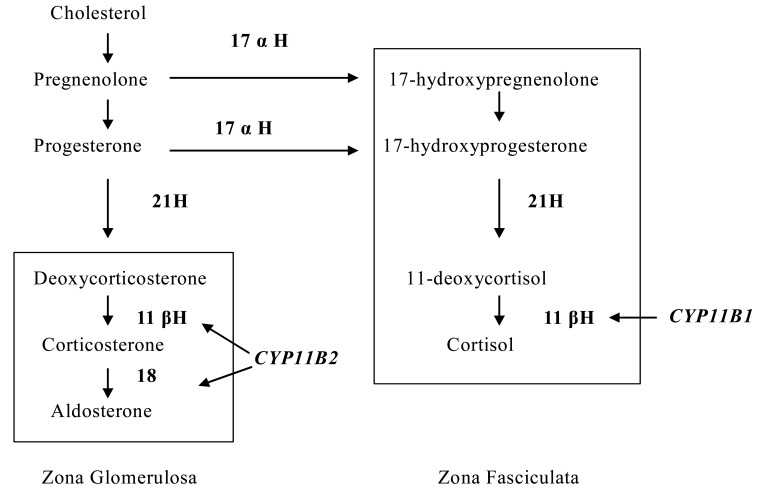 | Fig. 2Normal biosynthetic pathways for cortisol and aldosterone. 17α H, 17α-hydroxylase; 21H, 21-hydroxylase. 11βH, 11-β hydroxylase; 18, 18-hydroxylase/oxidase. [From Freel EM, Connel JNC : J Am Soc Nephrol 15:1993-2001, 2004]
|
Go to :
Cortisol metabolism
Circulating concentrations of cortisol are 100-1000-fold higher than those of aldosterone. Furthermore, cortisol has a high affinity for the MR. However, cortisol availability is modulated by two isoenzymes of 11β-hydroxysteroid dehydrogenase, 11β-HSD1 and 11β-HSD2. Both enzymes control the interconversion of biologically active cortisol to inactive cortisone. 11β-HSD1 is widely distributed but is most abundantly expressed in liver and fat tissue. Principally the enzyme has bi-directional activities, capable of carrying out both 11-oxoreductase (cortisone to cortisol) and dehydrogenase reactions (cortisol to cortisone). 11β-HSD2 is a unidirectional dehydrogenase inactivating cortisol to cortisone. 11β-HSD2 exhibits a distinct tissue-specific expression in classical mineralocorticoid target tissues, such as epithelial cells from the distal nephron, colon and salivary glands, where it serves to protect the MR from cortisol (Fig. 1). Thus, in vivo MR selectivity to aldosterone relies on the inactivation of cortisol to cortisone by 11β-HSD2 at the pre-receptor level. Aldosterone, in contrast to cortisol, is not metabolised by 11β-HSD2.
Following interconversion of cortisol and cortisone, both steroids undergo A-ring reduction by 5α- and 5β-reductases and 3α-hydroxysteroid dehydrogenase (3α-HSD) to yield 5β-tetrahydrocortisol (THF), 5α-tetrahydrocortisol (allo-THF) and 5β-tetrahydrocortisone (THE). Overall 11β-HSD activity in the body is reflected by the ratio of total urinary cortisol and cortisone metabolites: [THF+allo-THF]/THE (Fig. 3A)4).
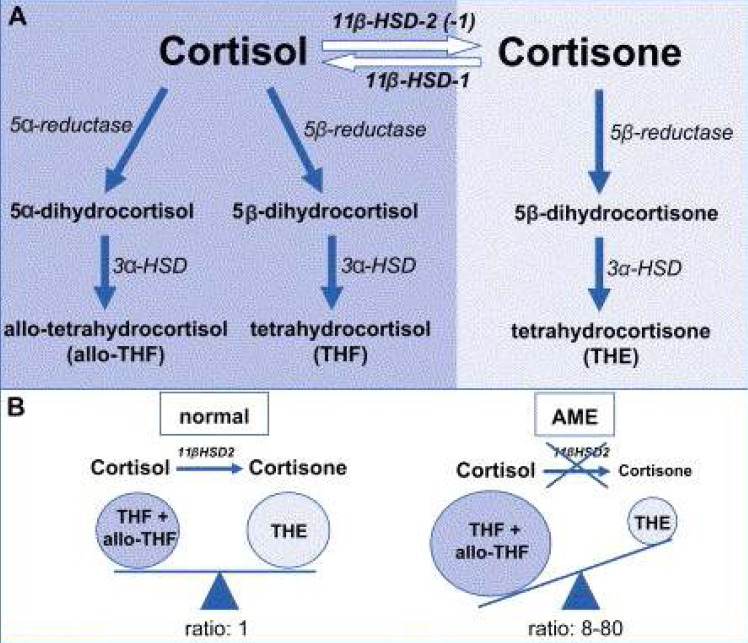 | Fig. 3A) Schematic depiction of the enzymatic activity involved in glucocorticoid metabolism. In the liver, 5α- and 5β-reductases and 3α-hydroxysteroid dehydrogenases (3α-HSDs) convert cortisol to 5α-tetrahydrocortisol (allo-THF) and 5β-tetrahydrocortisol (THF) and convert cortisone to tetrahydrocortisone (THE). B) In normal subjects, urinary excretion of cortisol metabolites compared to the cortisone metabolite is equivalent, resulting in a [THF+allo-THF]/THE ratio of 1. Mutations that inactivate 11β-HSD2 in apparentmineralocorticoid excess (AME) patients result in a grossly increased urinary excretion of THF and allo-THF compounds, whereas THE is dramatically reduced, resulting in a high [THF+allo-THF]/THE ratio. [From Hammer F, Stewart PM : Best Pract Res Clin Endocrinol Metab 20:337-353, 2006]
|
Go to :
Primary aldosteronism (PA)
Mineralocorticoid hypertension is a potentially reversible cause of hypertension that is characterized by the triad of hypertension, metabolic alkalosis, and hypokalemia (Table 2)5, 6).
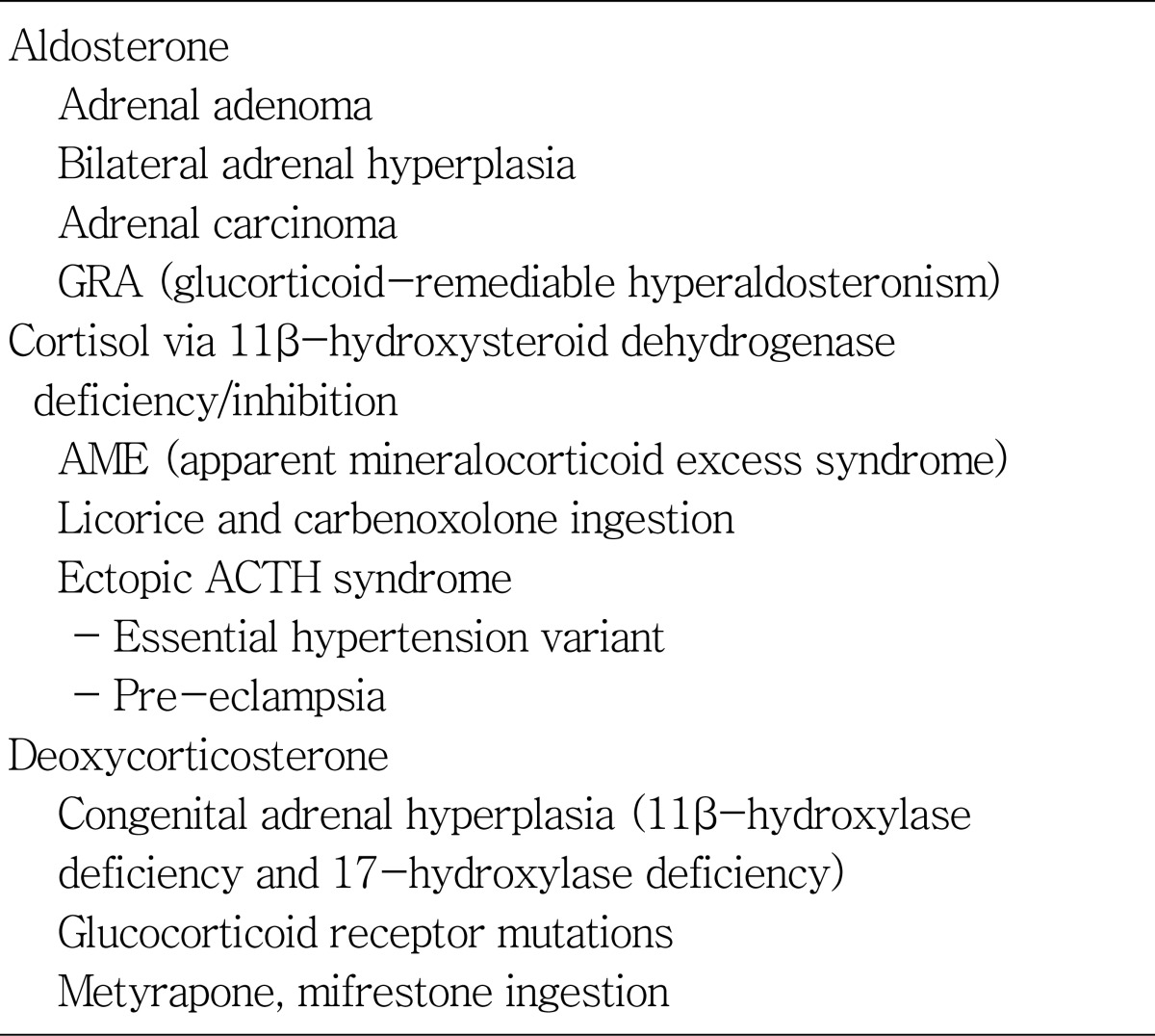
PA is the most common form of mineralocorticoid hypertension. The prevalence has varied from 0.5% to 10% of hypertensive patients. The two major subtypes are bilateral adrenal hyperplasia and adrenal adenoma. Familial forms of PA include familial hyperaldosteronism type 1 (FH-1) or glucocorticoid-remediable aldosteronism (GRA), and FH-2 (familial occurrence of bilateral adrenal hyperplasia, adrenal adenoma or both).
Screening for PA is a challenging topic. An important point is that not all patients with PA present with hypokalemia. Any of the following clues may prompt screening: (i) hypertension and unexplained persistent hypokalemia or diuretic-induced hypokalemia; (ii) severe hypertension (i.e. Joint National Committee 7 stage 2 hypertension, 160 mmHg systolic and/or 100 mmHg diastolic; (iii) patients with difficult-to-control hypertension requiring three or more drugs; (iv) when hypertension manifests at a young age (e.g. <20 years); (v) when adrenal incidentaloma is present; (vi) whenever an evaluation for secondary hypertension is considered; and (vii) hypertensive relatives of patients with PA.
The aldosterone renin ratio (ARR) has become the most widespread method of screening. Although there are no good data defining the optimal conditions and cut-off values of the ARR, an ARR 20 ng/dL per ng/mL per hour (554 pmol/L per ng/mL per hour) in combination with a plasma aldosterone concentration 15 ng/dL (416 pmol/L) in a morning blood sample from an ambulant patient with normokalemia is indicative of primary aldosteronism.
Once an abnormal ARR is seen then confirmatory test should be performed. The most widely used confirmatory tests for PA are intravenous saline loading, oral sodium loading and the fludrocortisone suppression test (FST). The captopril suppression test (CST) has also been introduced as a potential confirmatory technique for primary aldosteronism. Because the CST did not depend on sodium intake, it could be performed in patients for whom salt loading is contraindicated (such as those with cardiac failure) and is an efficient alternative test7). As treatment options differ for different forms of primary aldosteronism, it is important to determine the disease subtype. If biochemical confirmation is documented, high resolution adrenal computed tomography and/or magnetic resonance imaging should be performed. When the patient and clinician wish to pursue surgical treatment, adrenal vein sampling should also be performed in most patients.
Unilateral laparoscopic adrenalectomy is the optimal treatment for patients diagnosed with adrenal adenoma or unilateral hyperplasia. Patients with bilateral adrenal hypereplasia, and those with adrenal adenoma or unilateral hyperplasia who are not candidates for surgery or those who decline surgery, should receive a mineralocorticoid receptor antagonist. Two drugs of this class are currently available: spironolactone and eplerenone. Sprinolactone is more cost effective but has many unwanted antiandrogen and progesterone side effects. Substitution of the 17-thioacetyl group of spironolactone with a carbomethoxy group generates eplerenone. The affinity of eplerenone for progestin and androgen receptors is as much as 500-fold lower than that of spironolactone, thus nullifying the adverse effects associated with progesterone agonism and androgen antagonism.
Go to :
Familial forms of PA
GRA (FH-1) first reported in 1966 is the most common monogenic cause of human hypertension. GRA is an autosomal-dominant form of low rennin hypertension in which aldosterone excess is present under the influence of ACTH rather than angiotensin II. Subjects with GRA have two normal copies of genes encoding aldosterone synthase and 11β-hydroxylase, but they also have an abnormal gene duplication. This hybrid, or chimeric, gene combines the ACTH-responsive promoter sequence of the 11β-hydroxylase gene fused to the more distal aldosterone-synthase coding sequence. As a result, aldosterone synthase is ectopically expressed in the cortisol-producing zone of the adrenal cortex under the regulation of ACTH. The chimerism also results in the production of the hybrid steroids 18-oxocortisol and 18-hydroxycortisol that can be used as diagnostic aides8).
GRA is usually characterized by severe hypertension, sodium retention and suppressed plasma renin activity. Unlike other mineralocorticoid-excess states, hypokalemia in the absence of diuretic treatment is uncommon. The reason is unclear but may be related the influence of aldosterone on the circadian rhythms of ACTH release. A blunted aldosterone response to dietary potassium in GRA subjects may also explain the minimized potassium loss9). An important clinical feature associated with GRA is early hemorrhagic stroke and ruptured aneurysm, occurring at a mean age of 32 years and associated with high mortality (61%). As a result, screening with magnetic resonance imaging angiography, beginning at puberty and then every five years, has been recommended to detect asymptomatic intra-cranial aneurysms.
Diagnosis requires a high index of suspicion because GRA patients can have mild hypertension and are typically normokalemic; such patients are often misdiagnosed as having 'essential' hypertension. Clues pointing to a possible diagnosis of GRA include early onset of hypertension in youth (before age 20 years), a family history of early onset hypertension or early cerebral hemorrhage (before age 30 years), precipitation of hypokalemia when treated with potassium-wasting diuretics, and refractory hypertension refractory to standard treatments.
A confirmatory diagnosis can be made with either a dexamethasone-suppression test, measurement urinary 18/hydroxyl/oxy steroids, or direct genetic testing. In GRA the zona fasciculata ectopically produces aldosterone under the regulation of ACTH. As a result, when 0.5 mg of dexamethasone is given every 6 hours for two days, suppression of aldosterone to undetectable levels (<4ng/dL) is seen in GRA subjects10). GRA patients excrete large amounts of urinary 18-hydroxycortisol and 18-oxocortisol11) (socalled 'hybrid' steroids) reflecting the action of aldosterone synthase on cortisol in the zona fasciculata.
Treatment of GRA manipulates the dependency of aldosterone secretion on ACTH. Treatment with low dose glucocorticoids is effective. The typical dose for adults ranges form 0.125 mg to 0.25 mg dexamethasone, or 2.5 mg to 5 mg of prednisolone daily, usually administered at bedtime. The therapeutic goal should be normotension, and not normalization of biochemical markers, such as urinary 18-oxosteroid or serum aldosterone levels, since these remain elevated in the majority of patients whose blood pressure normalizes. The type I mineralocorticoid receptor antagonists, eplerenone and spironolactone, are effective treatment alternatives. Amiloride and triamterene, sodium-epithelial channel antagonists, have also been used successfully.
In clinical practice, FH-2 is indistinguishable from bilateral adrenal hyperplasia and adrenal adenoma, and the diagnosis can be made only by documenting primary aldosteronism in other family members and excluding GRA with genetic testing.
Go to :
Apparent mineralocorticoid excess syndrome (AME)
AME is an autosomal-recessive disorder characterized by clinical features suggesting excessive production of a mineralocorticoid-like substance with hypertension, plasma volume expansion, hypokalemic alkalosis, and a suppressed renin-angiotensin-aldosterone system. AME is the outcome of defective 11β-HSD212). In its full expression AME is rare, with fewer than 100 cases reported worldwide, but presentation is dramatic. Usually patients are children with low birth weight, failure to thrive, short stature, and severe, often fatal, hypertension with hypokalemic metabolic alkalosis and muscle weakness. Hypokalemic nephropathy sometimes causes nephrocalcinosis, polycystic kidney, and nephrogenic diabetes insipidus manifesting as thirst and polyuria. Renal insufficiency is not rare.
Biochemical abnormalities comprise suppressed PRA, undetectable serum aldosterone levels and hypokalemia. The diagnosis is based on urinary cortisol metabolites, which show a greatly increased [THF+allo-THF]/THE ratio of 8-80 (reference range 0.7-1.3) with very low or absent levels of THE clearly suggesting a defect in 11β-HSD2 activity (Fig. 3B)4). Interestingly, the excretion of 5α-cortisol metabolite s exceeds that of 5β-cortisol metabolites, resulting in a high urinary allo-THF/THF ratio, which suggests an additional defect in 5β-reductase activity. However, the [THF+allo-THF]/THE ratio provides only an overall index of 11β-HSD activity within the body. Thus, a high urinary free cortisol (UFF) to urinary free cortisone (UFE) ratio will be diagnostic, as it only reflects 11β-HSD2 activity in the kidney, but not 11β-HSD1 activity in the liver14, 15). DNA analysis is required to confirm the diagnosis.
Treatment involves blockade of the MR with a high dose of spirinolactone or eperlenone and potassium repletion. Patients have been successfully treated with the potassium sparing diuretics triamterene and/or amiloride. Suppression of cortisol with dexamethasone has had variable success. Thiazide diuretics are indicated when hypercalciuria and/or nephrocalcinosis are present. AME was reported "cured" in one patient following kidney transplantation due to the normal 11β-HSD2 activity of the transplanted kidney12).
An acquired form of AME can be seen with licorice ingestion. The active ingredient of licorice is glycyrrhizic acid, which is hydrolyzed into its aglicone glycyrrhetinic acid in vivo. A number of commercial preparations containing licorice are available such as herboristic and cosmetic; moreover some preparations are used as a cough remedy. A preparation of the root of the licorice plant (carbenoxolone) was successfully used to treat patients with peptic ulceration. Glycyrrhizic and glycyrrhetinic acids have a very low affinity for the MR, but are very potent competitive inhibitors of 11β-HSD216).
Go to :
Ectopic ACTH Syndrome
Cushing's syndrome can also present signs with signs and symptoms of mineralocorticoid excess, particularly in the sub-group caused by ectopic ACTH production. The ectopic ACTH syndrome is characterized by mineralocorticoid excess, with hypokalemic alkalosis found in 95-100% of cases, in contrast to <10% in other forms of Cushing's syndrome. ACTH has no direct effect on 11β-HSD2, but the enzyme is saturated in ectopic ACTH syndrome by very high concentrations of ACTH-dependent 11β-HSD substrates such as cortisol and corticosterone. Both the urinary ratio of THF+allo-THF/THE and UFF/UFE are elevated, not because of impaired 11β-HSD2 activity, but because of substrate saturation17).
Go to :
Liddle syndrome
Liddle syndrome is a rare form of an autosomal-dominant disorder. This syndrome results from constitutive activation of the epithelial Na channel (ENaC), resulting in sodium retention and urinary potassium and hydrogen ion wasting, causing hypokalaemic alkalosis with early onset severe hypertension and suppressed renin and aldosterone (due to low renin plus hypokalaemia)18).
The amiloride-sensitive ENaC is comprised of three subunit proteins (α, β and γ) having short cytosolic termini, 2 transmembrane domains, and a large extracellular loop. The α subunit appears to be required for the assembly or function of the whole complex. Liddle syndrome is caused by missense or frameshift mutations that delete or disrupt a proline-rich motif (PPPxY) in the intracytoplasmic C-terminal 'tails' of the β or γ subunits. These mutations increase expression of the ENaC at the cell surface by disrupting its binding to Nedd4-2, a ubiquitin-protein ligase that targets the ENaC for degradation19).
The condition should be suspected in patients (especially children) who present with hypertension and hypokalaemic alkalosis. It can be distinguished from hypertensive forms of congenital adrenal hyperplasia, primary glucorticoid resistance and AME by normal levels of 17α-hydroxyprogesterone, 11β-deoxycortisol and cortisol, normal ratios of free cortisol to cortisone (or metabolites) in urine and failure to respond to dexamethasone suppression of ACTH.
Although mineralocorticoid antagonist (spironolactone) is obsolete, K-sparing diuretics, specific ENaC inhibitor, are the drug of choice for patients with Liddle's syndrome. The use of amiloride and triamterene are enhanced by dietary salt restriction.
Go to :
Conclusion
The presence of mineralocorticoid excess should be suspected in any patient with the triad of hypertension, unexplained hypokalemia, and metabolic alkalosis. Avoiding hypokalemia is beneficial in several cardiovascular disease states including acute myocardial infarction, heart failure, and hypertension. In hypertensive patients, it seems beneficial to aim for serum potassium levels 3.5 to 5.0 mEq/L. All pharmacologically managed patients with mineralocorticoid excess should receive a mineralocorticoid receptor antagonist to prevent the negative effects of aldosterone on nonepithelial tissues.
Go to :
 XML Download
XML Download