

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Acute renal failure (ARF) is mainly caused by renal ischemia or nephrotoxic agents. It is characterized by severe reduction in glomerular filtration rate (GFR), urinary concentrating ability, and impaired tubular reabsorption of sodium. A diminished adenylate cyclase activity and aquaporin-2 (AQP2) expression may contribute to the urinary concentration defect in post-ischemic kidneys1). Several studies reported defects in the collecting duct sodium handling in ischemia/reperfusion (I/R)-induced ARF rats2, 3). Although the mechanisms underlying intrarenal vasoconstriction and hypoperfusion remain incompletely defined, an imbalance of vasoactive agents such as endothelin and nitric oxide may be contributory4). On the other hand, the tubular cell damage may be caused by such factors as ATP depletion, intracellular calcium, oxygen free radicals and apoptosis5).
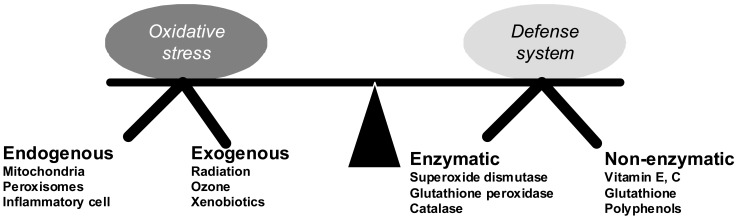
Under normal circumstances, the kidney generates reactive oxygen species (ROS), including superoxide anions, hydrogen peroxide, peroxynitrite and hydroxyl radical, which are efficiently eliminated by enzymatic-superoxide dismutase (SOD), catalase, glutathione peroxidase (GPX)-and non-enzymatic systems (glutathione, Vitamins C and E). Oxidative stress occurs when ROS production overrides metabolic capacity of the antioxidant defense system, often resulting in tissue damage (Fig. 1)6).
Numerous interventions have been put forward to counteract the effects of ROS, by reinforcing the antioxidant defense systems. Dietary supplementation with the antioxidant vitamin E slowed the rate of progression of renal deterioration7). Recently, α-lipoic Acid (α-LA) has been known as a potent antioxidant (with a redox potential E0+ of -290 mV compared with vitamin E, which has a redox potential E0+ of +370 mV), endogenously existing in tissues and acting as a cofactor of key mitochondrial enzymes, controlling glucose oxidation, such as the pyruvate dehydrogenase and the α-ketoglutarate dehydrogenase8). Its anti-oxidant effects are due to direct radical scavenging and metal chelation by both LA and its reduced form, dihydrolipoic acid (DHLA)9). It also has been demonstrated that LA reduces activation of nuclear factor (NF)-kb, a transcriptional factor in cultured endothelial cells and fractalkine-mediated inflammatory processes in endotoxemia10).
This review aims at providing recent insight into the role of α-LA in the regulation of AQP water channels and sodium transporters in acute renal injury caused by I/R injury and cisplatin-treatment.
Localization of AQP water channels and sodium (co)transporters
In the kidney, at least six isoforms of AQP proteins have been detected (AQP1, -2, -3, -4, -6, and -7). Among them, AQP1 is primarily responsible for the constitutive water permeability of proximal tubules and thin descending limbs of Henle11). Studies using AQP1-knockout mice revealed a dual role of AQP1 in proximal tubular reabsorption and medullary hypertonicity construction12). However, AQP2 to AQP4 are mainly expressed in the collecting duct. AQP2 is specifically located in the principal cells of the collecting duct and is regulated in the short term and long term via the arginine vasopressin (AVP)/cyclic AMP pathway13). In addition, the constitutive localization of AQP3 and AQP4 in the basolateral membrane of principal cells confers to the epithelium high water permeability, in concert with AQP2 insertion into the apical membrane14).
The tubular reabsorption of solutes is basically linked to the activity of Na,K-ATPase that is heavily expressed in the basolateral membrane throughout the nephron segments. On the contrary, the sodium transporters in the apical membrane differ in different tubular segments. In the proximal tubule, type-3 Na-H exchanger (NHE3) is apically expressed. On the other hand, the apically expressed bumetanide-sensitive Na-K-2Cl cotransporters (NKCC2) and NHE3 are mainly responsible for the sodium reabsorption in the thick ascending loop of Henle (TAL). In the distal convoluted tubule, the thiazide-sensitive sodium chloride cotransporter (NCC) is involved in the apical movement of sodium14-16).
Changes of AQP and sodium transporters in ischemia/reperfusion renal injury and cisplatin induced nephropathy
1. Ichemia/reperfusion renal injury
The urinary concentration defect and increased water excretion is, at least in part, accounted for by a reduced abundance of AQP2 water channels in the collecting duct1). Furthermore, cAMP generation in response to AVP was attenuated in ARF, most prominently in the outer medulla. Generation of cAMP in the outer medulla in response to forskolin was not affected, but sodium fluoride was significantly blunted in ARF. Therefore, the primary impairment may lie at the level of membrane G protein that acts as a transducer between AVP receptor and adenylate cyclase, decreasing the AVP-stimulated adenylate cyclase activity and hence subsequent generation of cAMP. We have also demonstrated that the abundance of several major renal sodium transporters (Na-KATPase, NHE-3, NKCC2, and NCC) was severely reduced following I/R injury, in association with an impairment of tubular reabsorption of filtered sodium. Decreased levels of sodium transporters along the nephron may play a role in the impairment of tubular sodium reabsorption.
Previous studies have shown that treatment with anti-inflammatory agents, significantly reduce the down-regulation of AQP-1,-2 and AQP-3 levels as well as major renal sodium transporters (NHE3, NaPi-2, BSC-1, TSC, and Na-K-ATPase) in rats with bilateral ischemia-induced ARF17, 18). The antioxidant may play key role against the glomerular inflammatory processes, through a diminution of the activity of inflammatory enzymes and cytokine secretion, or by inhibiting the activity of NF-kB19).
2. Cisplatin-induced nephropathy
Cisplatin is highly efficacious in treating a wide variety of neoplastic disease20). However, its clinical use is often limited by its potential renal toxicity leading to ARF21-23). The most silent and persistent features of cisplatin-induced nephropathy is polyuria and inability to concentrate urine24). We have demonstrated that cisplatin-treatment caused polyuric renal failure in association with decrease in free water reabsorption25). The expression of AQP1 and AQP2 was decreased in the cortex, outer medulla and inner medulla, whereas that of AQP3 was decreased in the outer medulla and the inner medulla. Immunohistochemistry also revealed that cisplatin decreased immunoreactivity for AQP1, AQP2, and AQP3. The AVP evoked generation of cAMP was attenuated by cisplatin, being most prominent in the outer medulla.
Effects of α-LA on the dysregulation of AQP and sodium transporters
ARF was ameliorated by treatment with α-LA at the time of ischemia26). Accordingly, the creatinine clearance was increased. Furthermore, α-LA treatment attenuated the polyuria that was encountered in the post-ischemic period, along with normally expressed abundance of AQP1-3. The reduced expression of NHE3 and Na,K-ATPase and the increase in FENa were completely prevented by treatment with α-LA. Overall, α-LA treatment 1) markedly inhibited the decline of GFR, 2) reduced the defects in urinary concentration and normalized the sodium handling, 3) prevented the down-regulation of AQP, being consistent with the improvement of urinary concentration ability and morphologic changes.
The underlying mechanism of α-LA treatment to prevent I/R injury or cisplatin-induced nephropathy remains to be established. It was shown that nitric oxide inhibits AVP-stimulated osmotic water permeability in the isolated and perfused cortical collecting duct of the rat kidney27). Yu et al28) demonstrated that nitric oxide production is stimulated by hypoxic injury in epithelial cells, and an inhibition of its synthesis protects against hypoxic injury in rat proximal tubule cells. Moreover, endothelin, and chemokines have been implicated in the urinary concentrating defect associated with ureteral obstruction, a condition which has been also associated with reduced expression of AQP1, AQP2, and AQP329, 30). These results suggest changes of renal hormones such as nitric oxide and endothelin may contribute to recovery renal function following α-LA treatment in I/R- or cisplatin induced nephropathy. However, its underlying molecular mechanisms are probably multifactorial, and remain to be further elucidated.
 XML Download
XML Download