

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
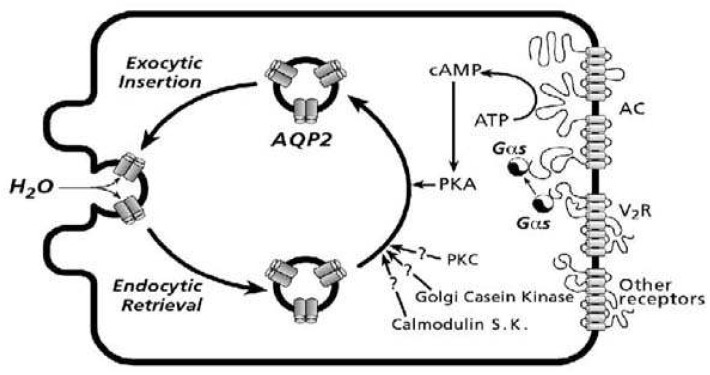
A major function of the kidney is to regulate body water and sodium balance. This function is achieved and finely regulated by a number of cellular and molecular processes, including tubular reabsorption of water and sodium through renal water channels (aquaporins) and sodium transporters under the tight control of hormones and nerves along with intracellular signaling pathways. Vasopressin and angiotensin II (AngII) are important peptide hormones that are chiefly involved in the renal conservation of water and sodium (1:2). Vasopressin controls body fluid homeostasis through the regulation of renal tubular water reabsorption. The action of vasopressin has been previously reviewed extensively1-5). Its main site of action in kidney tubules is the collecting duct, where vasopressin binds to the vasopressin V2-receptor and stimulates an increase in intracellular cAMP content via adenylyl cyclase. Subsequently, cAMP activates protein kinase A (PKA) which phosphorylates various proteins including aquaporin-2 (AQP2)6-8). In the collecting duct principal cells, AQP2 is then translocated from intracellular vesicles to the apical plasma membrane (Fig 1, 2)9), thereby increasing the osmotic water permeability. Moreover, transcriptional regulation of AQP2 in vivo is thought to be a result of vasopressin-induced increase in intracellular cAMP levels, which is capable of stimulating AQP2 gene transcription.
The renin-angiotensin-aldosterone system has also been demonstrated to play a critical role in the regulation of renal sodium and water metabolism through a variety of physiological pathways. In particular, AngII has known effects on the regulation of renal hemodynamics, glomerular filtration rate, aldosterone secretion, as well as more direct effects on renal tubule transport in the proximal tubule10). Moreover, several recent studies have demonstrated that AngII also has an important effect on the thick ascending limb (TAL) and collecting duct where AngII receptor mRNA and protein are present. For example, 1) AngII stimulates the activity of epithelial sodium channel (ENaC) in an isolated cortical collecting duct11); 2) AngII induces vasopressin V2-receptor mRNA expression in cultured inner medullary collecting duct (IMCD) cells12); and 3) AngII increases vasopressin-stimulated facilitated urea transport in the rat terminal IMCD13). These findings may suggest that AngII could play a role in the regulation of urinary concentration capacity. Moreover, we recently demonstrated that 1) AngII infusion in rats increased the expression of Na-H exchanger NHE3 and Na-K-2Cl cotransporter type 2 (NKCC2) in the medullary thick ascending limbs14); 2) pharmacological blockade of the AngII AT1 receptor in rats co-treated with dDAVP and dietary NaCl-restriction resulted in an increase in urine production, a decrease in urine osmolality, and blunted the dDAVP-induced upregulation of AQP215); and 3) AngII plays a direct role in the regulation of AQP2 short-term targeting to the plasma membrane in the IMCD cells through AT1 receptor16).
The action of vasopressin and AngII is mediated by intracellular secondary messengers, which are mainly coupled to the cAMP/PKA and phosphoinositide pathways, respectively. It is well known that vasopressin induces an increase in intracellular cAMP levels17), while AngII induces a rise in intracellular [Ca2+] by inositol 1,4,5-triphosphate (IP3) and protein kinase C (PKC) activation by diacylglycerol18). Interestingly, several previous studies demonstrated evidence of cross-talk between the signaling pathways of vasopressin and AngII. For example, AngII potentiates the vasopressin-dependent cAMP accumulation in Chinese hamster ovary (CHO) cells transfected with cDNA of both AT1A receptor and V2-receptor19). Moreover, forskolin potentiates AngII-induced increase in intracellular [Ca2+] in an isolated cortical TAL20). Consistent with this, we demonstrated that pharmacological blockade of the AngII AT1 receptor in rats cotreated with dDAVP and dietary NaCl-restriction (to induce high plasma endogenous AngII levels) resulted in an increase in urine production, a decrease in urine osmolality, and blunted the dDAVP-induced upregulation of AQP2 and phosphorylated-AQP2 (p-AQP2: AQP2 phosphorylated in the PKA-phosphorylation consensus site Ser-256) expression in the inner medulla15).
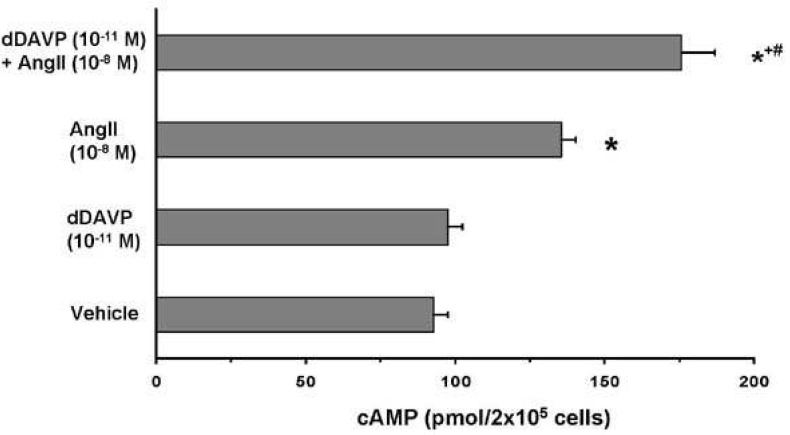
Regulation of intracellular AQP2 trafficking and osmotic water permeability in the collecting duct principal cells involves intracellular cAMP production21). Accordingly, we also demonstrated that vasopressin and AngII have additive effects on intracellular cAMP accumulation in primary cultured IMCD cells (Fig 3), stimulation of AQP2 phosphorylation, and AQP2 trafficking; hence, it could increase the osmotic water permeability of the collecting duct cells. To directly address this, we examined the direct effect of 1) single treatment with either dDAVP or AngII and 2) co-treatment with dDAVP AngII + in primary cultured IMCD cells. Moreover, we examined the effect of single treatment with dDAVP or AngII in AQP2-transfected Madin Darby Canine Kidney (MDCK) cells. These subjects are discussed in detail in the following sections which were based on the previously published data from our labs14-16, 22).
Occupation of AngII AT1 receptor plays a role in regulation of vasopressin-regulated AQP2 expression in collecting ducts and urine concentration in vivo.
Urinary concentration and dilution depends on the presence of a discrete segmental distribution of transport properties along the renal tubule and urinary concentration depends on 1) a hypertonic medullary interstitium, which is generated by active NaCl reabsorption as a consequence of countercurrent multiplication in water-impermeable nephron segments; 2) a high water permeability (constitutive or vasopressin-regulated) in other renal tubular segments for osmotic equilibration, which chiefly depends on the expression of aquaporins; and 3) flow rate of tubule fluid entering the medullary collecting ducts. The collecting duct represents the final site for the control of water excretion into urine. Water permeability of the collecting duct is tightly regulated, under the control of the vasopressin, which causes a dramatic increase in collecting duct water permeability, allowing reabsorption of water from the tubular fluid down an osmotic gradient. We demonstrated that long-term dDAVP treatment upregulates inner medullary AQP2 and p-AQP2 expression, whereas cotreatment with the AT1 receptor antagonist candesartan prevented the increase in the expression of inner medullary AQP2 and p-AQP2 in response to the long-term dDAVP treatment15). On the contrary, outer medullary NKCC2, NHE3, and Na,K-ATPase expression, which are chiefly involved in the active reabsorption of NaCl by the medullary thick ascending limb to maintain the countercurrent multiplication, was unchanged in response to dDAVP treatment, neither in the presence nor the absence of candesartan15). This suggests that the progressive blunting of the urine concentration in response to dDAVP seen in the rats co-treated with AT1 receptor blocker candesartan was primarily due to the prevention of a dDAVP-mediated increase in the osmotic water permeability in the outer and inner medullary collecting ducts, despite long-term dDAVP treatment. This is evidenced by the maintained (not increased) expression of AQP2 and p-AQP2, respectively.
dDAVP or AngII has a stimulatory effect on AQP2 targeting to the plasma membrane in vitro
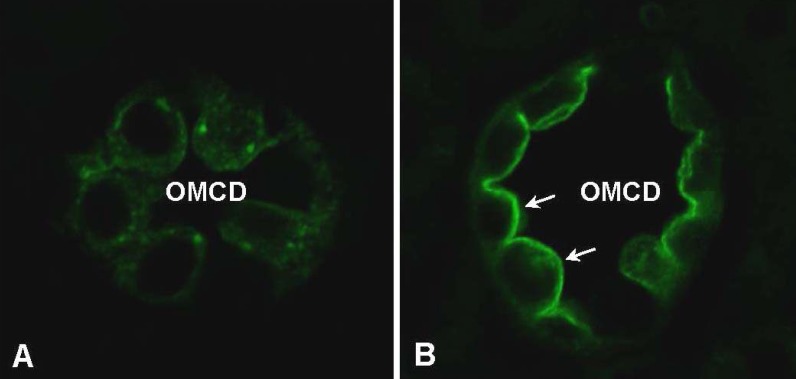
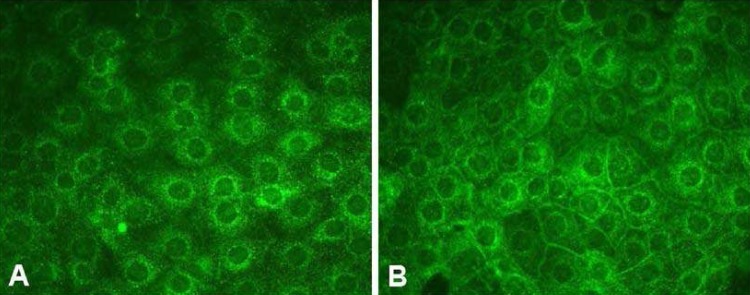
We demonstrated that AQP2 targeting to the plasma membrane of primary cultured IMCD cells was enhanced in response to acute dDAVP treatment, consistent with a number of previous in vitro and in vivo studies8, 23-25). Interestingly, we demonstrated that short-term treatment of AngII was also associated with increased AQP2 targeting to the plasma membrane in IMCD cells (Fig. 4) and this was inhibited by co-treatment with the AngII AT1 receptor blocker candesartan. This suggests that short-term treatment of AngII could, at least partly, play a role in the activation of signal transduction pathways leading to AQP2 recruitment to the plasma membrane and that this was mediated by AngII AT1 receptor activation.
The signal transduction pathways for AQP2 targeting to the plasma membrane have been investigated in a number of previous studies4). Cyclic-AMP levels in the collecting duct principal cells are increased in response to vasopressin binding to the V2-receptor and subsequently, cAMP activates protein kinase A (PKA) which phosphorylates various proteins including AQP27). Consistent with this, we demonstrated that cAMP levels increased in response to dDAVP in a dose-dependent manner in primary cultured IMCD cells16). Cyclic-AMP levels were also significantly increased by the single treatment of AngII and importantly, co-treatment with dDAVP+ AngII revealed an additive effect on cAMP production (Fig. 3), p-AQP2 expression, and AQP2 targeting in primary cultured IMCD cells16). It should be noted that we demonstrated enhanced AQP2 targeting to the plasma membrane and increased cAMP levels in response to treatment of AngII, alone, with no significant changes in the p-AQP2 expression at 15 min after administration16). Since intracellular cAMP accumulation in IMCD cells is known to activate PKA, which subsequently increase AQP2 phosphorylation and AQP2 trafficking, an increase in p-AQP2 expression would be expected. However, at the time point used no significant changes were observed but it cannot be excluded that at some time point during the stimulation AQP2 phosphorylation may have been increased. Moreover, it remains possible that only a minor fraction of AQP2 is subject to phosphorylation and at a level where fractional change escapes detection. Phosphoproteomic analysis of kidney tissues after dDAVP treatment is currently undertaken to examine the course of changes of the dDAVP-induced phosphorylated proteins.
Co-treatment with dDAVP and AngII potentiates the cAMP production, p-AQP2 xpression, and AQP2 targeting in vitro
The actions of peptide hormones, vasopressin, and AngII, are mediated by intracellular secondary messengers. Vasopressin induces an increase in intracellular cAMP levels, while AngII induces a rise in intracellular [Ca2+] by IP3 and PKC activation by DAG. Our findings revealed that AngII had an additive effect on the dDAVP-induced cAMP production (Fig 3), p-AQP2 expression, and AQP2 targeting16). This suggests cross-talk between these intracellular signaling pathways of these two peptide hormones, compatible with our findings obtained from in vivo studies revealing that AngII AT1 receptor blockade decreased dDAVP-induced water reabsorption and AQP2 levels in the rat kidney15).
How does short-term treatment of AngII stimulate the cAMP accumulation, AQP2 phosphorylation, and AQP2 targeting in the primary cultured IMCD cells? It is known that both an increase in the intracellular [Ca2+] and the cAMP levels are required for the AQP2 targeting and the accompanying increase in osmotic water permeability23, 26). The role of Ca2+ in the cross-talk between AngII and vasopressin was investigated in a previous study19) using CHO cells transfected with cDNA of both the AT1A receptor and the V2-receptor. This study demonstrated that chelation of intracellular Ca2+ lowered the cAMP accumulation by vasopressin and markedly decreased the potentiation of vasopressin-induced cAMP accumulation by AngII19). On the other hand, the same study also demonstrated that increased intracellular [Ca2+], induced by thapsigargin comparable to that induced by AngII, did not potentiate the vasopressin-dependent cAMP accumulation19), indicating that increase in intracellular [Ca2+] does not likely play a major role for the cross-talk. Consistent with this, Lorenz et al.27) demonstrated that AQP2 shuttling is evoked neither by the AVP-dependent increase of [Ca2+] nor AVP-independent increase of [Ca2+] in primary cultured IMCD cells from the rat kidney. However, it should be mentioned that there is a discrepancy between the results from primary cultured IMCD cells27) and isolated perfused IMCD tubules23) with regard to the role of intracellular [Ca2+] in vasopressin-induced AQP2 trafficking. This might be related to the expression levels of vasopressin receptors and/or AQP2, or other elements in the AQP2 trafficking system and further studies are needed to clarify this discrepancy.
Another transduction pathway of AngII is PKC. We demonstrated that cAMP levels were significantly increased in response to short-term AngII treatment; whereas, the levels were not increased in response to AngII treatment in the presence of the PKC inhibitor staurosporine16). Moreover, imunocytochemistry demonstrated that AQP2 targeting to the plasma membrane was stimulated by AngII treatment alone, whereas it was attenuated in response to AngII+ staurosporine16). These findings suggest that an activation of PKC could, at least in part, play a role in stimulation of the adenylate cyclase and AQP2 targeting in the primary cultured IMCD cells. AngII-mediated intracellular signaling is not restricted to [Ca2+] and PKC, but AngII-mediated activation of ERK, JNK, and STAT also exists. In particular, ERK, p38 kinase, and phosphatidylinositol 3'-kinase (PI3-kinase) were shown to regulate AQP228). Further studies are, therefore, warranted to examine whether AngII plays a role in the long-term regulation of AQP2 by increasing AQP2 gene transcription by activation of MAP kinase and PI3-kinase, as demonstrated by insulin28).
Aldosterone increases urine production and decreases apical AQP2 expression in rats with diabetes insipidus
In addition to angiotensin II, aldosterone also plays a role in the regulation of body fluid homeostasis22, 29, 30). Aldosterone has a complex interaction with vasopressin and this has been the subject in several previous studies31, 32). Synergistic effects of mineralocorticoids on vasopressin-induced osmotic water permeability have been shown in the toad urinary bladder and isolated perfused cortical collecting duct (CCD) from rabbits, but not in rats31, 32). Interestingly, in congenitally vasopressin-deficient Brattleboro rats, mineralocorticoid-deficiency (adrenalectomized Brattleboro rats substituted with glucocorticoids) causes a decreased diluting capacity compared to Brattleboro rats with intact adrenal glands, while the urine flow was not markedly affected33). Moreover, pharmacological interference with the renin-angiotensin system in Brattleboro rats using either captopril (an angiotensin converting enzyme inhibitor) or spirononlactone (a mineralocorticoid receptor blocker) has been shown to reduce urine production34, 35), suggesting that aldosterone may play a role for regulation of water excretion in vivo in the absence of vasopressin.
Recently, we demonstrated a marked increase in urine production in response to aldosterone treatment of rats with lithium-induced nephrogenic diabetes insipidus and in vasopressin-deficient Brattleboro rats with central diabetes insipidus22). Importantly, aldosterone treatment was associated with impaired or decreased apical trafficking of AQP2 in the connecting tubule (CNT) and CCD22). In addition to the decreased apical AQP2 expression in both the CNT and the initial CCD (iCCD), increased AQP2 expression was seen in the basolateral domains of the iCCD22). Consistent with this, aldosterone treatment was associated with increased free water clearance in both lithium-treated rats and Brattleboro rats. In contrast, spironolactone and aldosterone receptor antagonist treatment was associated with decreased urine production and an apparent increase in the AQP2 labelling in the apical plasma membrane domain, suggesting that the aldosterone receptor could be involved in the regulation of AQP2 trafficking and urine production in conditions of diabetes insipidus22).
Moreover, we demonstrated that long-term aldosterone infusion in normal rats receiving water intake ad libitum was associated with polyuria and decreased urine concentration, accompanied by decreased apical but increased basolateral AQP2 labeling intensity in the CNT and CCD despite the absence of changes in AQP2 protein expression in the kidney cortex and outer medulla (unpublished data). The increased basolateral AQP2 in the CNT and CCD by long-term aldosterone infusion was not directly related to altered levels of plasma angiotensin II or low plasma potassium level (unpublished data). In contrast, short-term aldosterone infusion in rats did not alter the intracellular distribution of AQP2 in the CCD. Thus, these studies could indicate that aldosterone is, at least in part, involved in the regulation of body water balance through affecting the intracellular AQP2 distribution in the CNT and CCD in addition to the sodium reabsorption.
 XML Download
XML Download