

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Congenital adrenal hyperplasia (CAH) is caused by defects in various enzymes necessary for cortisol synthesis. 90-95% of CAH cases are caused by 21-hydroxylase deficiency (21-OHD). In patients with 21-OHD, poor synthesis of cortisol results in chronic stimulation of the adrenal cortex. This results in over-production of cortisol precursors. Some of these precursors are shunted into the androgen synthetic pathway, causing signs and symptoms of androgen excess, including ambiguous genitalia in females and rapid somatic growth with accelerated skeletal maturation in both sexes1).
Since the discovery of cortisone in the 1950s, CAH has been managed effectively, and the clinical course of the disease has markedly improved2). Typical medical treatment consists of oral glucocorticoid and mineralocorticoid administration in order to compensate for adrenal steroid deficiencies and to suppress adrenal androgens.
Herein, we report four cases of patients who were diagnosed as having CAH. They have been managed with hydrocortisone and fludrocortisone. But the patients stopped taking medicine without the doctor's consent. Thereafter, adrenal masses were found by abdominal computed tomography (CT) scan in all four cases. Recent reports reveal a high apprearance of small tumors in patients with untreated CAH and most adrenal masses in children with CAH are benign3-5). Two of our four cases were managed with adrenalectomy because of increasing adrenal mass size and virilization. The size of the other two cases did not increase. So they were treated conservatively without adrenalectomy.
Case Report
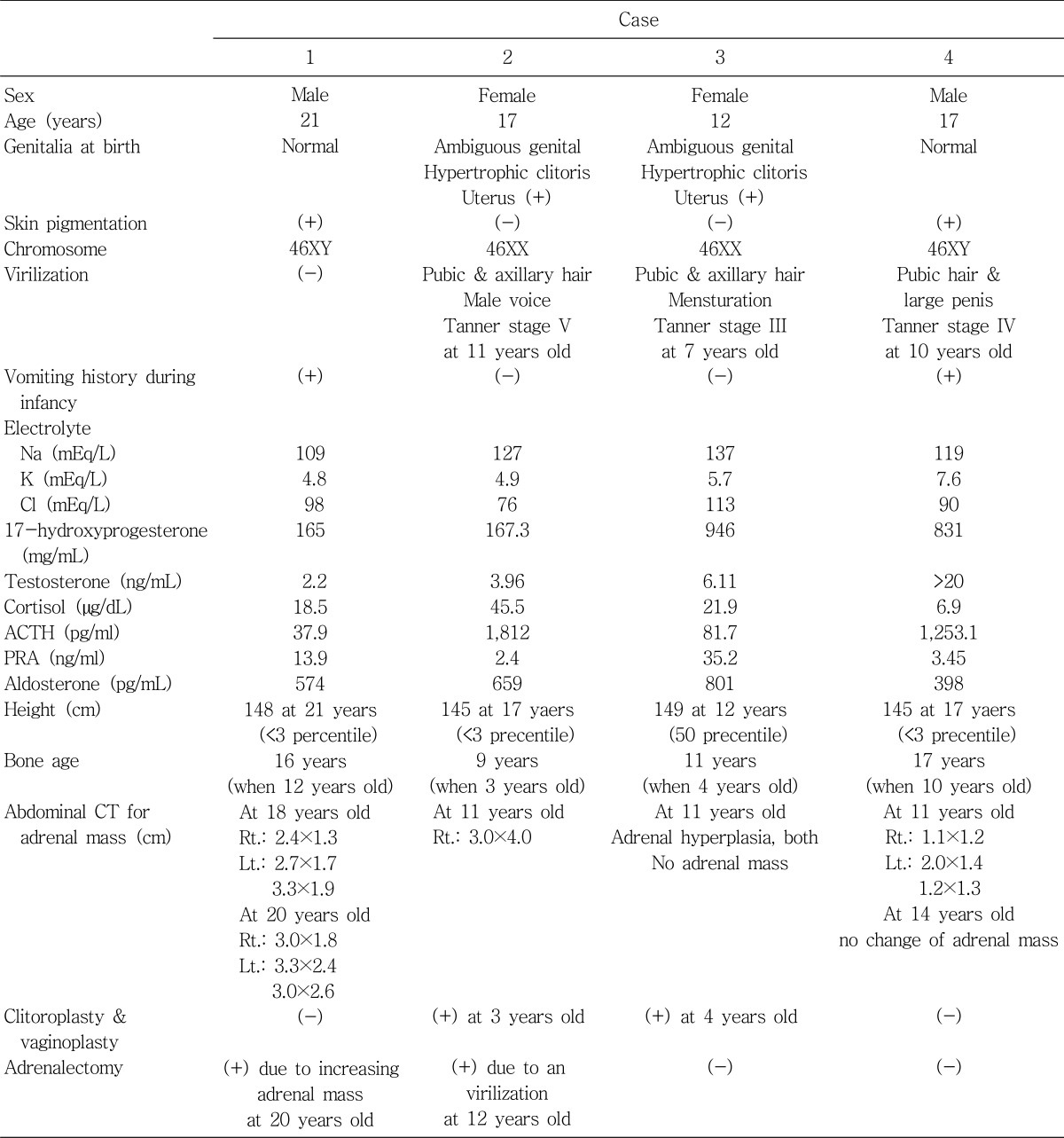
Clinical features of 4 patients are shown in Table 1.
1. Case 1
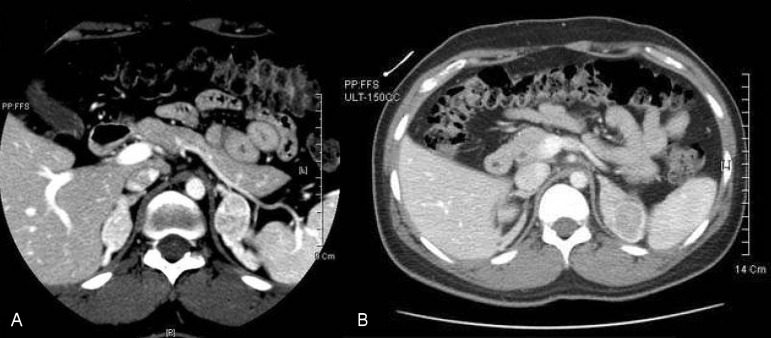
A male neonate who presented with severe vomiting at 15 days of life was brought to medical attention. He was first admitted to a local hospital where his symptoms improved. However severe vomiting recurred after he was discharged and he was brought to Kyunghee medical center. Laboratory evaluation revealed a pH level of 7.36, HCO3- 13 mEq/L, base excess -10 mEq/L, and Na+ 109 mEq/L. He was diagnosed as salt-losing 21-OHD CAH. Hydrocortisone and fludrocortisone therapy had begun. Electrolytes were normalized and vomiting was resolved. Thereafter the level of Na+ was normalized. However at the age of 20 years, his height was 145 cm (less than 3 percentile). Abdominal CT performed at the age of 18 years (July 30, 2004) revealed the presence of multiple bilateral adrenal masses (possibly adenoma). The size of his right adrenal mass was 2.4×1.3 cm and the size of left adrenal masses were 2.7×1.7 cm and 2.3×1.9 cm with adrenal hyperplasia. Follow-up abdominal CT was performed at the age of 20 years (Fig. 1). Abdominal CT showed that both adrenal masses on both sides were increasing in size. The size of his right adrenal mass was 3.0×1.8 cm, and the size of his left adrenal masses were 3.3×2.4 cm and 3.0×2.6 cm. Bilateral adrenalectomy was performed at the age of 20 years.
2. Case 2
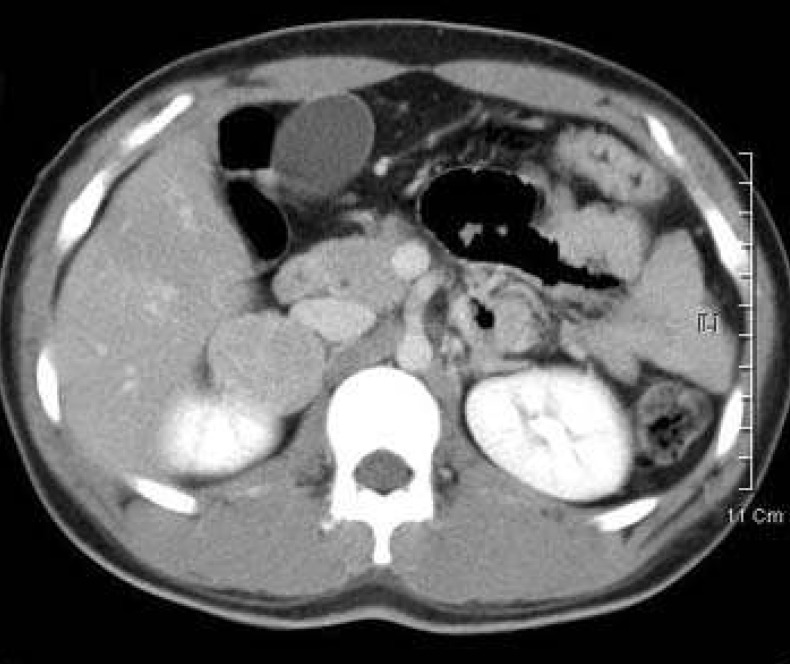
A female neonate presented with ambiguous genitalia at birth suggesting salt-losing CAH. Hydrocortisone and fludrocortisone were initiated. When she was at the age of 3 years, vaginoplasty and clitoroplasty were performed. At the age of 11, she showed breast budding and pubic hair. Masculine voice and virilization were also present. Abdominal CT performed at the age of 11 years showed a 3.5×4.0 cm mass in her right adrenal gland (Fig. 2). Adrenal mass was assumed to be an adrenal adenoma. Right adrenalectomy was performed at the age of 12 years.
3. Case 3
A female neonate showed ambiguous genitalia at birth. She was diagnosed as having 21-OHD CAH. Hydrocortisone and fludrocortisone administration was carried out during follow-up and vaginoplasty and clitoroplasty were performed at the age of 4 years. When she was 11 years old and 12 years old, abdominal CTs were performed due to her virilization including hirsuitism and masculine voice. They revealed adrenal hyperplasia without change in size. There was no remarkable finding of her adrenal mass.
4. Case 4
A male neonate was admitted to medical attention at 1 month of age with poor oral intake and diarrhea. He was diagnosed with renal tubular acidosis type IV. He was discharged after his oral intake and diarrhea improved. He was admitted to Kyunghee medical center at 5 months of age with poor oral intake and vomiting. Laboratory evaluation revealed a pH level of 7.35, HCO3- 14 mEq/L, base excess -9 mEq/L, and Na+ 119 mEq/L. During follow-up, he was diagnosed with CAH. Hydrocortisone and fludrocortisone medication was initiated. After his medication had begun, the level of Na was normalized. During follow-up, subdural hematoma and lymphangioma over his right calf developed and was surgically removed. Medication was stopped on his parents' request and since the age of six, he showed secondary growth such as hyperpigmentation, hirsuitism, large penis, and pubic hair. Abdominal CT performed at the age of 14 years revealed the presence of adrenal masses (Left adrenal gland : 2.0×1.4 cm & 1.2×1.3 cm, Right adrenal gland : 1.1×1.2 cm). Since there was no change in the size of adrenal mass, adrenalectomy was not performed.
Discussion
CAH describes a group of autosomal recessive condition in which deletions or mutations of the cytochrome P450 21-hydroxylase gene result in glucocorticoid and/or mineralocorticoid deficiency6). In 95% of cases, 21-OHD is responsible for the disease7). Here, epidemiology, genetics, pathophysiology, clinical features, and the management of CAH will be reviewed as follows.
Data from several neonatal screenings show that CAH due to 21-OHD is not uncommon. Incidence varies according to ethnicity and geographical area. The overall incidence of classical 21-OHD is 1:10,000-1:15,000 in most reported populations8).
The 21-hydroxylase gene CYP21A2 is located on chromosome 6p21.3 within the HLA histocompatibility complex9). Two highly homologous 21-hydroxylase genes exist; CYP21A2 and CYP21A1P, of which CYP21A2 codes for the active 21-hydroxlyase and CYP21A1P is an inactive pseudogene. More than 90% of mutations of the active CYP21A2 gene are generated by recombinations between the active and the inactive pseudogene10). Proximity to the HLA histocompatibility complex is a cause of high rate of recombination.
The pathophysiology of CAH due to 21-OHD is closely linked to the degree of enzyme deficiency6). The biosynthesis of cortisol is regulated by negative feedback on hypothalamic corticotropin-releasing hormone (CRH) and pituitary adrenoconticotrophic hormone (ACTH) secretion. A defect in cortisol biosynthesis leads to a compensatory increase in hypothalamic and pituitary secretion of CRH and ACTH. This stimulates the adrenal cortex and results in the pathological correlative of hyperplastic adrenal glands. Cortisol and aldosterone biosynthesis are compromised because of an insufficient enzymatic conversion.
The consequence of 21-OHD is a state of hypocortisolism, hypoaldosteronism, and hyperandrogenism combined with catecholamine deficiency and several metabolic disturbances. Metabolic defects result in insulin resistance, metabolic syndrome, and infertility6). Endogenous glucocorticoids are essential for the development and continuing regulation of the adrenal medulla, synthesizing epinephrine and norepinephrine. Without cortisol, the organogenesis of the adrenal medulla is severely disturbed and the consequence is catecholamine deficiency, mainly comprising a lack of epinephrine11).
CAH is classified as classic, the severe form, or non-classic, the mild or late-onset form. Classic CAH is subclassified as the salt-losing or non-salt-losing (simple virilizing). Non-classic CAH is subclassified as the late-onset form or cryptic form. Salt-losing CAH is a severe form with a concurrent defect in aldosterone biosynthesis. Non-salt-losing CAH is a form with apparently normal aldosterone biosynthesis. Non-classic CAH may be asymptomatic or may be associated with signs of androgen excess developing during childhood or at puberty.
The classic forms are characterized by a congenital decrease of cortisol and aldosterone secretion and an increase in androgen biosynthesis. All female infants with classic CAH due to 21-OHD typically have ambiguous genitalia at birth because of exposure to high concentrations of androgens in the uterus. The internal genitalia are normal, including uterus, fallopian tubes, and ovaries; Wolffian duct structures are not present. Boys with 21-OHD may show completely normal external genitalia. Boys with classic CAH have no signs of CAH at birth, except subtle hyperpigmentation and possible penile enlargement. Thus, the age at diagnosis in boys varies according to the severity of aldosterone deficiency. Up to 2/3 of patients suffering from classic 21-OHD have insufficient aldosterone biosynthesis leading to salt-losing episodes after birth and later on12). Boys with the salt-losing form typically present at 7-14 days of life with vomiting, weight loss, lethargy, dehydration, hyponatraemia, hyperkalaemia, and can present shock.
Patients with non-classic CAH do not have cortisol deficiency, but instead have manifestations of hyperandrogenism, generally later in childhood or in early adulthood13, 14). There is no prenatal virilization. Postnatal presentation usually becomes obvious in the peri-pubertal period. Manifestations include premature pubarche, tall stature, advanced bone age, menstrual irregularities, infertility, hirsutism and acne.
CAH due to 21-OHD can be diagnosed in newborn screening programs by screening for 17-hydroxyprogesterone in dried blood spots. Today, most cases of classical 21-OHD are diagnosed with this tool. A high concentration of 17-hydroxyprogesterone in a randomly timed blood sample is diagnostic of classic 21-OHD.
The gold standard for diagnosis of CAH is a corticotropin stimulation test, with measurement of 17-hydroxyprogesterone at 60 min. A stimulated concentration of 17-hydroxyprogesterone higher than 45 nmol/L is diagnostic of 21-OHD.
The aim of medical treatment in 21-OHD is to suppress adrenal androgens sufficiently without impairing growth, while allowing for normal pubertal development and fertility.
In classic CAH, glucocorticoids are given in doses sufficient to suppress excessive secretion of CRH and ACTH without total suppression of the hypothalamic-pituitary-adrenal. Physiological cortisol secretion rates are about 6-8 mg/m2/day15-17). To achieve these goals, the glucocorticoid doses have to exceed the physiological cortisol secretion rate, using doses of 12-18 mg/m2 hydrocortisone daily divided into two or three doses10). Hydrocortisone is the glucocorticoid of choice during childhood18).
Mineralocorticoid replacement is achieved with fludrocortisone. Fludrocortisone was administered in cases of salt-losing manifestation and elevated plasma renin activity, to decrease glucocorticoid doses, or according to the genotype19). The use of fludrocortisone in patients with non-salt-losing classic CAH allows management with lower doses of glucocorticoid and improves linear growth18-20). Infants with the salt-losing form of 21-OHD require mineralocorticoid (fludrocortisone, usually 0.1-0.2 mg) and sodium chloride supplements (1-2 g daily; 1 g NaCl contains 17 mEq of sodium) in addition to glucocorticoid treatment.
Many patients with the non-classic CAH do not need treatment. Treatment is indicated only for those with symptoms and aims to reduce hyperandrogenism18-20).
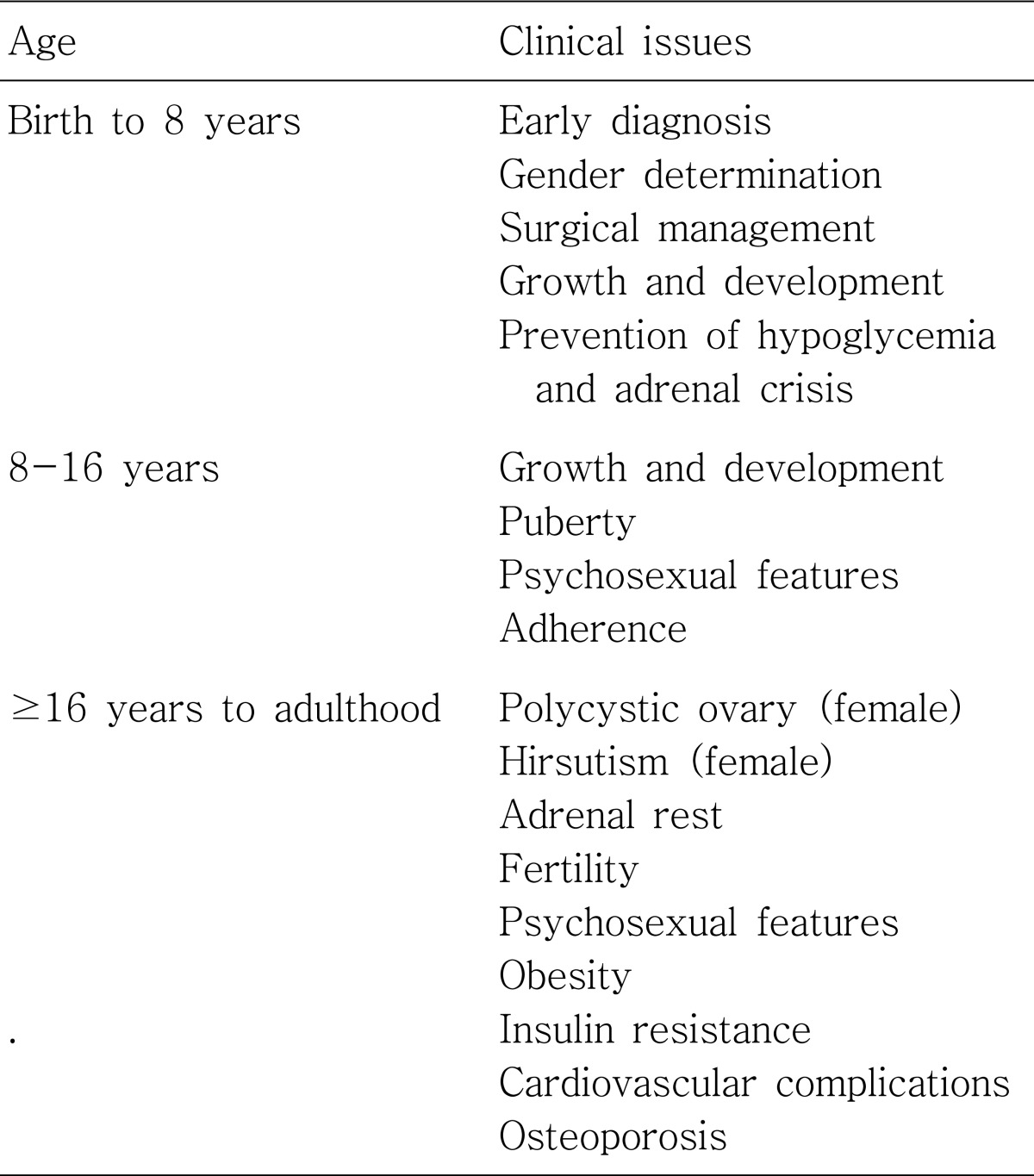
CAH constitutes a continuum of disorders that affect patients throughout their lives (Table 2). A variety of clinics involving close interaction between paediatric, reproductive, adult endocrinologists, and clinical psychologists experienced in psychosexual counselling will be necessary for patients with CAH21-24).
The development of adrenal adenoma or carcinoma in patients with CAH is rare; the etiology is not clear. However, the incidence of adrenal masses appears to be higher in CAH patients and in heterozygotes than in the general population3). Recent reports have revealed a high prevalence of small adrenal tumors in patients with untreated 21-OHD CAH3, 4). Histological types of adrenal tumor include adenoma, myelolipoma, and hemangioma25). Rarely, virilizing adrenal carcinoma has been found in patients with CAH, but most adrenal masses in children with CAH are benign5). The pathogenesis of adrenal tumor in patients with 21-OHD CAH is thought to be related to a consequence of ACTH hypersecretion, which results from the lack of glucocorticoid synthesis26). Avoidance of high steroid dose is likely to result in insufficient suppression of corticotropin stimulation27). The importance of compliance with medications is emphasized. Chronic poor compliance to therapy appears to be associated with the development of the tumor. Therefore patients with 21-OHD CAH should maintain steroid medication to avoid the appearance of adrenal tumor. Follow-up imaging may help to discover the appearance of adrenal tumor in patients with long-term management of 21-OHD CAH.
There has been much improvement during the past 50 years in the understanding of CAH, and the management with CAH continues to improve. Furthermore new medical strategies that offer the prospect of an improved outcome continue to evolve. However, patients with 21-OHD CAH should maintain steroid medication to avoid the appearance of adrenal tumor, to improve its symptoms, and to normalize electrolyte disorder, particularly hyponatremia.
 XML Download
XML Download