

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Cyclosporine is a lipophilic cyclic peptide of 11 amino acids, while tacrolimus is a macrolide antibiotic. Both drugs have been isolated from fungi and possess similar suppressive effects on cell mediated and humoral immune responses. Patients treated with the calcineurin inhibitors are at high risk of developing renal injury1). Calcineurin inhibitor nephrotoxicity includes acuteazotemia, chronic progressive renal disease, and tubular dysfunction. Although renal insufficiency induced by calcineurin inhibitors has received the most attention, tubular dysfunctions are also clinically important and will be briefly reviewed in this article. Calcineurin inhibitor-associated tubular dysfunction is manifested by metabolic acidosis, hyperkalemia, calcium, phosphate wasting, and magnesium loss.
Metabolic acidosis
Metabolic acidosis is common in patients with kidney transplantation. The first case report with post-transplant renal tubular acidosis was described by Massry et al. almost three decades ago2). Renal tubular acidosis (RTA) is non-anion gap metabolic acidosis and is generally mild and a symptomatic in kidney recipients3). The reported prevalence of calcineurin inhibitor-associated RTA is 13-17% intransplanted patients4-6).
Calcineurin inhibitor-associated RTA can be both proximal and distal RTA. The former form of acidosis is characterized by bicarbonate wasting due to the toxic effects of calcineurin inhibitors. In contrast, distal or type IV RTA is characterized by the inability to excrete hydrogen ions6).
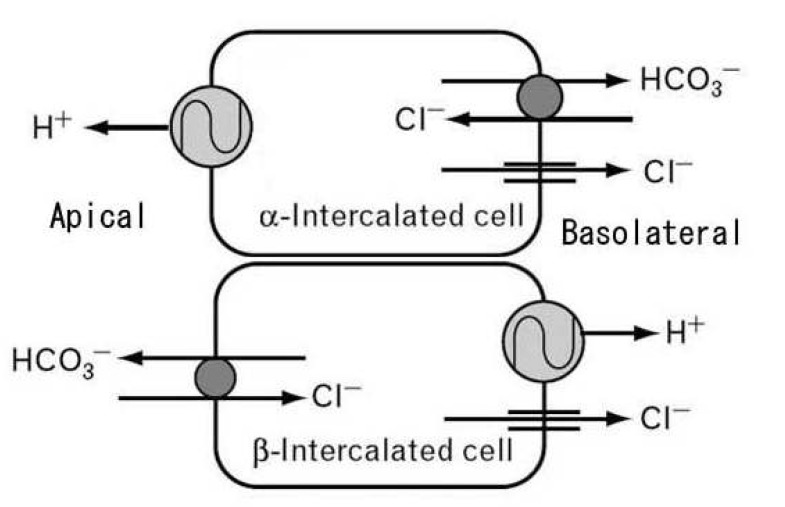
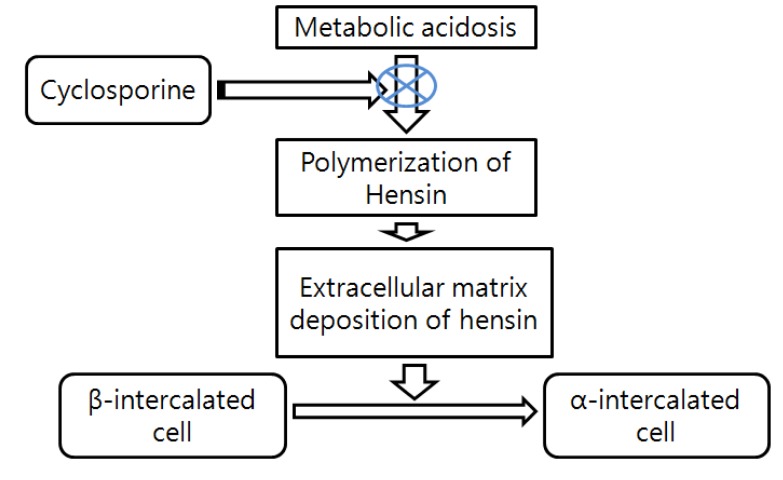
Use of calcineurin inhibitor cyclosporine can frequently cause type 4 RTA, a mild hyperchloremic acidosis, sometimes with elevated potassium. This may be reflecting decreased aldosterone activity and suppression of ammonium excretion by hyperkalemia7). There are some reports now that provide some insight as to how that might occur. Collecting ducts have 2 types of intercalated cells - the acid or hydrogen ion-secreting alpha-intercalated cells and the bicarbonate-secreting beta-intercalated cells (Fig. 1). The preponderance of which cell dominates rests on the type of diet. An acid diet leads to more alpha-intercalated cells, whereas an alkaline diet leads to more expression of beta-intercalated cells. These cells can interconvert according to the acid-base status. It has been reported that protein hensin is actually important in mediating transformation between beta-and alpha-intercalated cells. Metabolic acidosis normally induces the polymerization of the extracellular protein called hensin8). Deposition of hensin leads to the conversion of bicarbonate-secreting beta-intercalated cells into the acid-secreting alpha-intercalated cell. FK506 and cyclosporine inhibit polymerization of hens in protein. Consequently acid-secreting cells will be less abundant and risk for amild normal anion gap metabolic acidosis will increase8, 9)(Fig. 2). Aldosterone resistance is usually responsible for hyperkalemia induced by calcineurin inhibitor10, 11).
Hyperkalemia
Hyperkalemia is a recognized complication of cyclosporine and tacrolimus. Reported incidence of hyperkalemia is 5-40% among calcineurin inhibitortreated patients13, 14).
An elevation in plasma potassium concentration due to reduced efficiency of urinary potassium excretion is common in calcineurin inhibitor-treated patients. It may be severe and potentially life-threatening with concurrent administration of an angiotensin converting enzyme inhibitor or angiotensin receptor blocker. Cyclosporine may reduce potassium excretion by altering the function of several transporters, decreasing the activity of the renin-angiotensin-aldosterone system, and impairing tubular responsiveness to aldosterone15, 16).
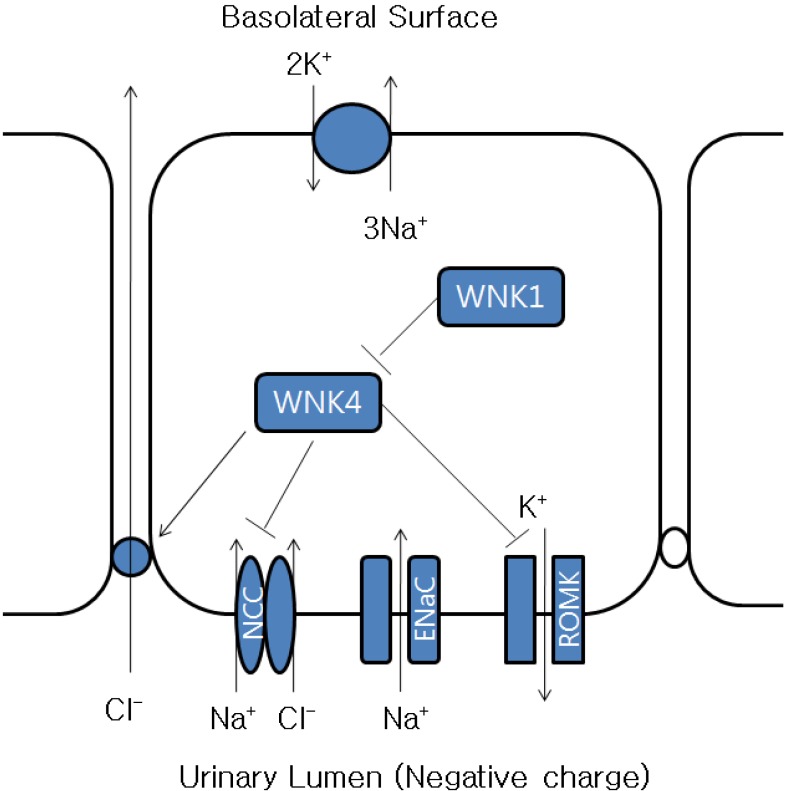
Renal potassium excretion is primarily derived from potassium secretion in the distal cortical nephron via potassium channels in the luminal membrane. This process is stimulated by sodium reabsorption, aldosterone, and the basolateral Na+,K+-ATPase pump. In vitro studies suggest that cyclosporine may directly impair the function of the potassium secreting cells in the cortical collecting tubule by affecting each of these steps: reduced activity of the Na+,K+-ATPase pump17-19), inhibition of the luminal potassium channel20), and increased chloride reabsorption16). Sodium reabsorption creates a lumen-negative electrical gradient that promotes potassium secretion. Cyclosporine increases paracellular or transcellular chloride reabsorption, which prevents generation of lumen-negative potential that drives potassium secretion. Calcineurin inhibitors inhibit luminal potassium channels and increase chloride reabsorption via alteration of WNK kinases21)(Fig. 3).
Cyclosporine may also have a secondary effect on potassium homeostasis in patients concurrently treated with a beta-blocker. In this setting, there is often a modest and transient elevation in the plasma potassium concentration due to movement of potassium out of cells into the extracellular fluid22). Why this occurs is not known.
Treatment is similar to that of hyperkalemia in chronic kidney disease: reduction in potassium intake, adjustment in medications, and so forth. One exception occurs in the early posttransplant period, in which renal insufficiency also contributes to the impairment in potassium excretion. Administration of a cation exchange resin increases the risk of intestinal necrosis at this time23). Hydration and a low-potassium diet are preferred during that period.
Hypophosphatemia
Urinary phosphate wasting induced by calcineurin inhibitors can cause hypophosphatemia. Cyclosporine inhibits the type IIa sodium phosphate cotransporter and probably the type IIb sodium phosphate cotransporter of the intestine24).
Many patients have hypophosphatemia during the first 6 months after transplant. This is due to increased urinary excretion of phosphate and probably decreased intestinal absorption. Post-transplant hypophophatemia is usually asymptomatic, but can rarely be severe enough to cause severe muscle weakness, including weakness of respiratory muscle. Phosphate supplementation should be prescribed for severe or symptomatic cases.
Hypomagnesemia
The calcineurin inhibitors cause hypomagnesemia by suppressing reabsorption of magnesium from renal tubules. Several case reports suggest that hypomagnesemia may contribute to calcineurin inhibitor-induced encephalopathy25-27). Hypomagnesemia has also been implicated as a contributor to the nephrotoxicity associated with cyclosporine28).
Hypercalciuria
It is known that cyclosporine treatment induces high bone-turnover osteopenia and hypercalciuria. Both cyclosporine and tacrolimus are associated with hypercalciuria. Animal studies suggest that hypercalciuria induced by cyclosporine administration is associated with an inhibition of calbindin D28k expression, resulting in calcium transport defect in the distal tubule29).
Conclusion
In conclusion, attention must also be paid to the calcineurin inhibitor associated renal tubular dysfunction such as renal magnesium wasting, calcium and phosphate wasting, distal tubular acidosis, and impaired renal potassium excretion, in addition to the well known side effects, such as tubulointerstitial fibrosis and glomerular or vascular damage.
 XML Download
XML Download