

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
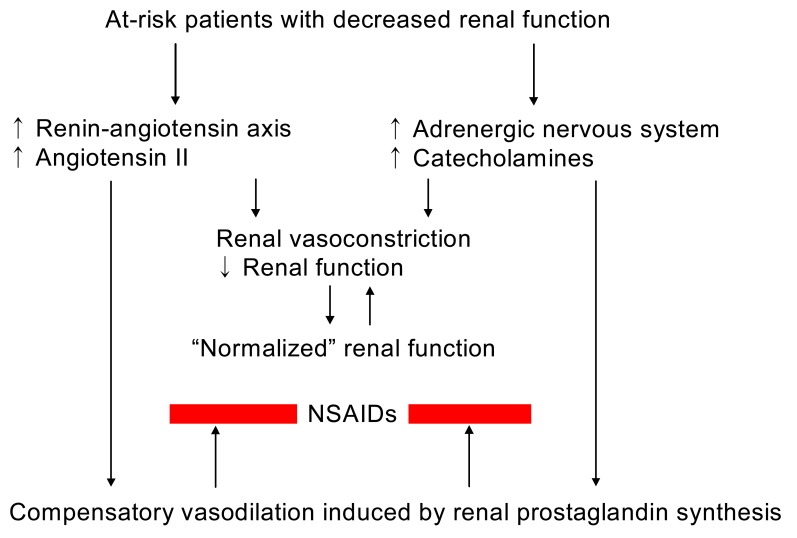
In the clinical conditions of decreased renal perfusion, such as nephritic syndrome, liver cirrhosis, heart failure, volume depletion, or aging kidneys, renal prostaglandin (PG) production mediated primarily by cyclooxygenase-1 (COX-1) and possibly by clooxygenase-2 (COX-2) plays a major role in maintaining renal hemodynamics. Inhibition of the synthesis of renal PGs by conventional non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit both COX-1 and COX-2, may result in acute renal complications such as sodium and water retention, hyperkalemia, acute renal failure, and acute papillary necrosis, or chronic renal effects such as nephrotic syndrome, chronic papillary necrosis, and analgesic nephropathy1-3). NSAIDs may also influence blood pressure in treated hypertensive individuals. The COX-2 specific inhibitors were first thought to have much less renal adverse effects by sparing homeostatic COX-1 activity in the kidney. However, emerging evidence is disappointing at this point and indicates that the cardiorenal risks associated with the use of COX-2 specific inhibitors and conventional NSAIDs are comparable. Regardless of cyclooxygenase specificity, up to approximately 5% of individuals exposed to NSAIDs may develop one or more forms of renal adverse effects, although the incidence of nephrotoxicity associated with the use of NSAIDs is relatively low in the normal population. Regarding the consumption of these compounds which account for approximately 2.5% of all prescription dollars and the populations that use NSAIDs frequently (old age and who have underlying diseases), the number of at-risk individuals should be high (Fig. 1)4).
This review will summarize the possible mechanisms by which fluid and electrolyte abnormalities, acid-base disturbances, and acute renal failure associated with NSAIDs use evolve.
Go to : 
Cyclooxygenase, prostaglandins, and kidney
PGs play active roles in the local regulation of vascular tone and salt and water homeostasis by modulating the glomerular hemodynamics and also by adjusting the function of the distal nephron5). In the early 1990s, the existence of two isoforms of cyclooxygenase (COX-1 and COX-2) was confirmed6). COX-1 isoform, which is expressed constitutively in many tissues and catalyzes the PG synthesis, is believed to have physiologic functions including gastric mucosal defense and platelet aggregation. COX-1 is abundantly expressed across species in the collecting ducts, renal vasculature, glomeruli, and papillary interstitial cells7). In contrast, basal COX-2 expression in the kidney is less apparent and exhibits some inter-species differences in its localization. COX-2 is constitutive in some tissues and is markedly induced by bacterial endotoxins, cytokines, and growth factors and catalyzes the synthesis of pro-inflammatory PGs.
In addition to their role in preserving renal perfusion, renal PGs promote the secretion of renin, impair sodium reabsorption in the loop of Henle and cortical collecting tubule, and partially antagonize the effect of antidiuretic hormone to increase water reabsorption in the collecting tubules1, 8). Locally generated PGs may also mediate part of the natriuretic effect of dopamine and of one of the atrial natriuretic peptides9).
These roles of PGs are not very important in normal subjects in whom basal renal PG production is relatively low. However, they may become important when prostaglandin synthesis is stimulated by underlying renal disease or by vasoconstrictors such as angiotensin II or norepinephrine, the secretion of which is increased in states of effective volume depletion; true volume depletion due to gastrointestinal or renal losses (as with diuretic therapy), heart failure, or cirrhosis (Fig. 2).
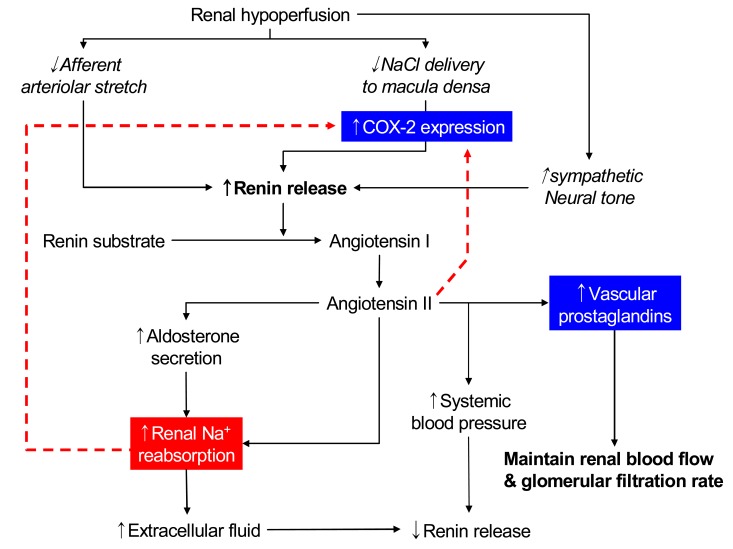
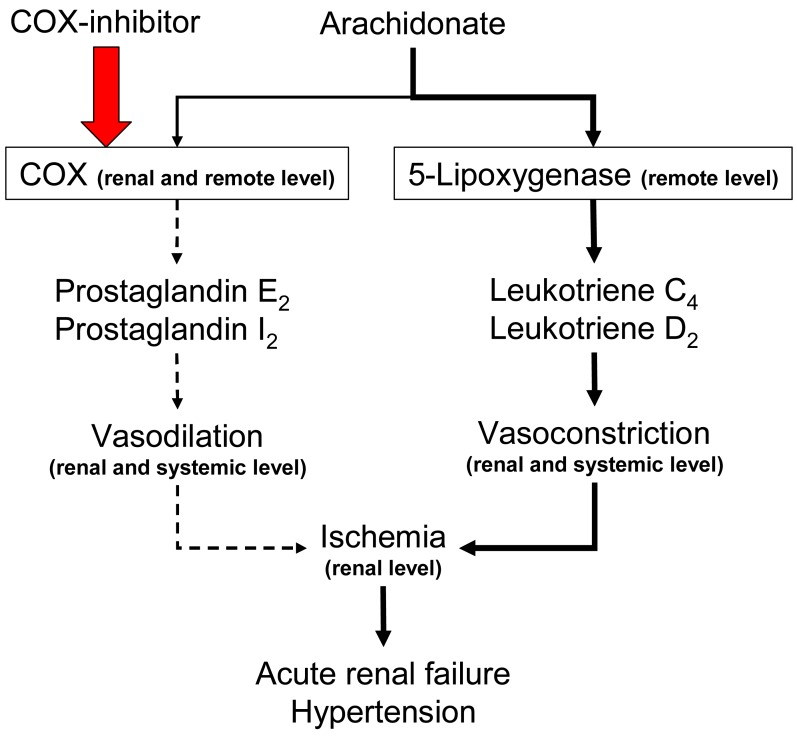
NSAIDs inhibit cyclooxygenase, impairing transformation of arachidonic acid to prostaglandins, prostacyclin, and thromboxanes, although the extent of enzyme inhibition varies among the different NSAIDs. Administration of cyclooxygenase-inhibiting NSAIDs in the setting of volume depletion interferes with local vasodilator effects of PGs and may result in a catastrophic decline in glomerular filtration rate (GFR), resulting in overt renal failure.
Go to :
Prostaglandin E2 and Epithelial solute and water transport
Prostaglandin (PGE2) either stimulates or inhibits epithelial solute and water transport along the nephron10). When added to vasopressin-prestimulated collect ing ducts, PGE2 potently inhibits water absorption11). At least three distinct effects of basolateral PGE2 on transport have been described: stimulation of basal water reabsorption; inhibition of vasopressin-stimulated water reabsorption; and inhibition of sodium reabsorption.
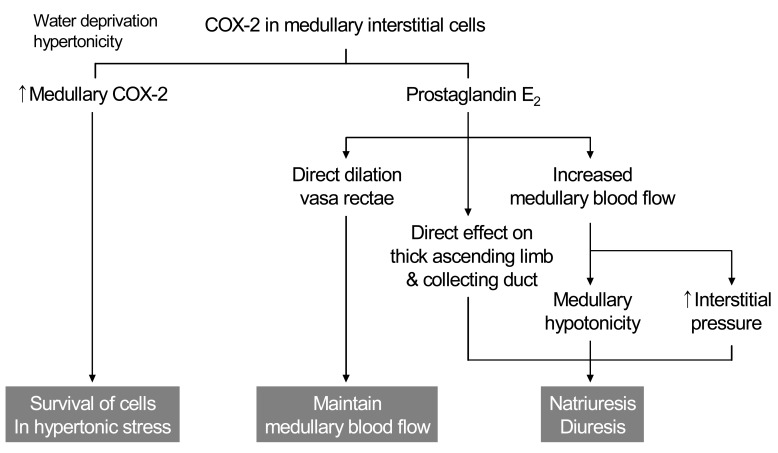
The mechanism by which PGE2 contributes to natriuresis may involve changes in resistance of the renal medullary microcirculation. Although expression of COX-2 in the macula densa and cortical thick ascending limb (cTAL) is increased by low salt diet, medullary interstitial COX-2 expression is increased by high salt diet. Increased PGE2 directly dilates the descending vasa recta and increases medullary blood flow, which may contribute to increased interstitial pressure and medullary hypotonicity, leading to enhanced salt excretion (Fig. 3).
PGE2 directly inhibits salt and water absorption in the in-vitro microperfused thick ascending limb (TAL) and collecting duct cells12), of which mechanisms are still uncertain. PGE2 directly inhibits chloride absorption in mouse or rabbit medullary TAL from either the luminal or basolateral surfaces. PGE2 also inhibits hormone-stimulated cyclic adenosine monophosphate (cAMP) generation in TALs. Because cAMP stimulates TAL transport, inhibition of cAMP generation through a Gi-coupled PGE2 receptor likely contributes to the inhibitory effects of PGE2 on TAL transport. In the collecting duct, PGE2 inhibits both vasopressin-stimulated osmotic water absorption and cAMP generation13). Intrarenal infusion of PGE2 is natriuretic, and although its effects on intrarenal hemodynamics undoubtedly play an important role, direct effects of PGE2 on epithelial transport are equally important. In contrast to PGE2-mediated inhibition of vasopressin-timulated water reabsorption, its capacity to inhibit sodium reabsorption is insensitive to pertussis toxin. Instead, PGE2 inhibits collecting duct sodium reabsorption via a calcium-dependent mechanism13).
Go to :
COX-2 Expression in the renal medulla
The renal medulla is a major site of prostaglandin synthesis and abundant COX-1 and COX-2 expression. The two cyclooxygenase isoforms exhibit different expression within the medulla, with COX-1 predominating in the medullary collecting duct cells and COX-2 predominating in medullary interstitial cells14). After dehydration, renal medullary COX-2 mRNA and protein expression are significantly induced, primarily in medullary interstitial cells15). In contrast, COX-1 expression in the mouse kidney is unaffected by water deprivation. Although hormonal factors may also contribute to COX-2 induction, shifting cultured renal medullary interstitial cells to hypertonic media (using either NaCl or mannitol) is sufficient to induce COX-2 expression. Osmotic induction of COX-2 expression in medullary interstitial cells plays an important role in the survival of these cells in hypertonic milieu after dehydration8).
Cultured medullary interstitial cells produce abundant PGE2, which has been shown to directly dilate vasa rectae, counteracting the constricting effect of angiotensin II and endothelin and thereby helping to maintain renal medullary blood flow16). Regulation of renal medullary blood flow has significant implications for regulating salt excretion and systemic blood pressure as previously described. Reduced medullary interstitial pressure increases renal sodium reabsorption17) and could attribute to sodium retention in the setting of NSAIDs use. Medullary interstitial prostaglandins may modulate epithelial solute and water reabsorption not only via hemodynamic effects, but also through direct effects on epithelial sodium reabsorption by the thick ascending limb and collecting duct. Loss of the tonic inhibitory effect of COX-2-derived PGE2 on salt absorption by these segments may contribute to the sodium retention seen with NSAIDs18).
Go to :
Cyclooxygenase-2 and the renal renin-angiotensin system
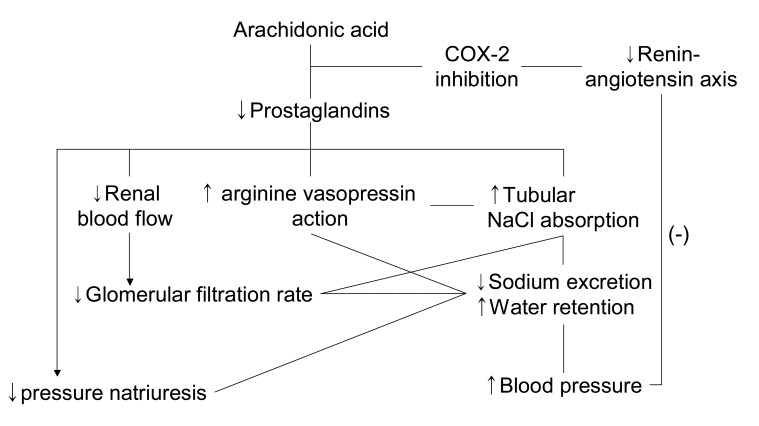
In the kidney, COX-2 is expressed in the macula densa/cTAL and medullary interstitial cells. The macula densa is involved in regulating afferent arteriolar tone and renin release by sensing alterations in luminal chloride concentration via changes in the rate of Na+-K+-2Cl- cotransport. Administration of non-specific cyclooxygenase inhibitors bluntly increases renin release mediated by macula densa sensing of decreases in luminal NaCl. High renin states such as salt deficiency, angiotensin converting enzyme (ACE) inhibitors or angiotensin receptor blockers, diuretic administration or experimental renovascular hypertension, are associated with increased macula densa/cTAL COX-2 expression. Furthermore, angiotensin II and/or aldosterone inhibits COX-2 expression19). In angiotensin II type 1 receptor knockout mice, COX-2 expression is increased similar to increases seen with ACE inhibitors or angiotensin II type 1 receptor blockers.
In vivo studies in rats indicated that increased renin release in response to low-salt diet, ACE inhibitor, loop diuretics, or aortic coarctation could be inhibited by administration of COX-2 inhibitors. In mice with genetic deletion of COX-2, ACE inhibitors or low-salt diet failed to increase renal renin expression, although renin was significantly increased in wild type mice. In contrast, in COX-1 null mice there were no significant differences in either the basal or ACE inhibitor-stimulated levels of renal renin activity from plasma or renal tissue compared with wild type mice.
By inhibiting the production of PGs that contribute to maintaining vasodilation of adjacent afferent arterioles, COX-2 inhibition may contribute to the decline in GFR observed in patients taking NSAIDs or selective COX-2 inhibitors. In healthy human subjects, COX-2 inhibitors may have minimal effects on renal hemodynamics. However, COX-2 inhibitors significantly decrease GFR in salt-depleted subjects. Further evidence of an important role for COX-2 in regulation of renal hemodynamics and renin production, acute renal failure, and hyperkalemia/type IV renal tubular acidosis (RTA) has been reported as acute nephrotoxic effects of COX-2 inhibitors, especially in the elderly.
Go to :
Adverse renal effects of NSAIDs
In the settings of enhanced PGs production, inhibition of PGs synthesis with NSAIDs could, in selected patients, lead to various electrolytes, acid-base disturbances such as hyponatremia, sodium retention, hyperkalemia, or renal tubular acidosis, which are usually reversible with discontinuation of the therapy. This is mediated in part by reduction in the release of renin and aldosterone, which also occurs with selective COX-2 inhibitors. In addition, vasoconstriction due to decreased synthesis of vasodilator PGs can exacerbate underlying hypertension and heart failure.
1. Sodium excretion
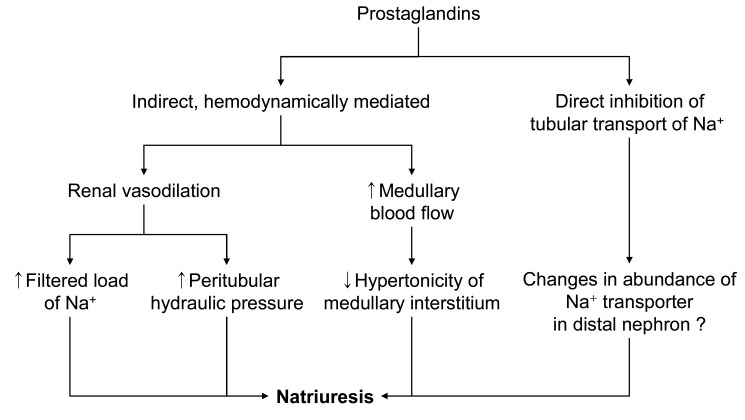
Renal PG has a natriuretic effect via reduced sodium reabsorption in the thick ascending limb and in the collecting tubules. Its effects on daily regulation of sodium balance are negligible in the basal state, but PGs play significant roles in hypovolemic states. In the setting of enhanced PG synthesis, NSAIDs could promote sodium retention and limit the response of edematous patients to diuretic therapy. PGs in the medulla may help to protect the thick ascending limb cells against ischemic injury when the patient is volume depleted. By diminishing NaCl reabsorption in the thick ascending limb, PGs could reduce the energy requirement of these cells, thereby allowing them to better tolerate the decrease in oxygen delivery. In the volume contracted state, PGs promote the release of renin from the kidney in response to loss of intravascular volume. And PGs counter the vasoconstriction effect of angiotensin II to maintain renal perfusion in the hyper-renin state.
2. Hyponatremia
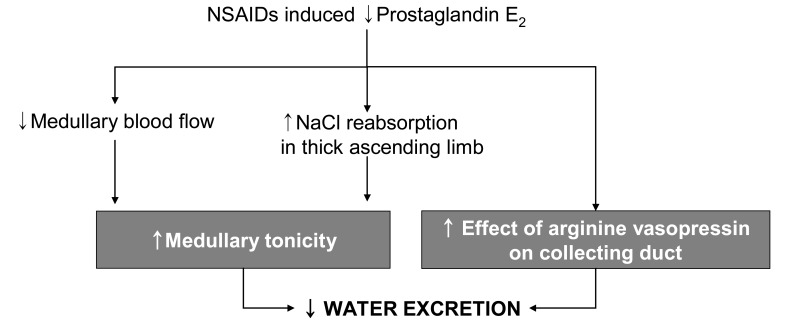
Intrarenal PGs may inhibit vasopressin-activated adenylcyclase activity in collecting duct epithelial cells, thus attenuating vasopressin-dependent transtubular water movement. Endogenously produced PGE2 in the kidney has an inhibitory effect on thick ascending limb cAMP levels and, based on the role of cAMP to increase Na+-K+-2Cl- cotransporter expression in the thick ascending limb, provides a tonic inhibitory influence on Na+-K+-2Cl- cotransporter expression20). PGs may also increase medullary blood flow, thus lowering medullary interstitial osmotic driving forces. Therefore, a PG-dependent, complex, intrarenal short-loop feedback, as well as possible extrarenal mechanisms, may be involved in the renal regulation of water balance (Fig. 4)21). Removal of the inhibitory effect of PGs on antidiuretic hormone (ADH) activity can diminish free water excretion21), of which cellular mechanisms is still unclear. The ensuing water retention can promote the development of hyponatremia when there are high and relatively nonsuppressible levels of ADH due to syndrome of inappropriate secretion of antidiuretic hormone (SIADH) or effective volume depletion. NSAIDs may also increase the susceptibility of elderly subjects to thiazide diureticnduced hyponatremia. The incidence of clinically symptomatic hyponatremia associated with NSAIDs use is very low. Exercise-associated hyponatremia has been widely researched and there is strong evidence that overhydration with hypotonic fluids is the primary cause. Concern has been expressed over the possible causative role of NSAIDs in exercise associated hyponatremia, which is still inconclusive22-24).
3. Edema
Administration of NSAID to human subjects removes the PGs' inhibitory effect on sodium and water reabsorption, leading to ADH induced water reabsorption and elevation of urine osmolarity that can exceed 200 mOsm/kg H2O, and causes reduction in plasma Na+ concentration, especially in volume depletion or SIADH (Fig. 5). In normal subjects, the initial fall in plasma Na+ will diminish ADH secretion, minimizing water retention. Thus, normal subjects may gain 0.5 to 1 kg, but the degree of sodium retention is more prominent in patients with underlying heart failure or cirrhosis. The increase in sodium transport contributes to the relative resistance to diuretics seen with NSAIDs therapy. Sodium retention leading to peripheral edema has also been observed with the selective COX-2 inhibitors.
In review of the currently available clinical data, it becomes apparent that the intrarenal functional role of COX-2 is predominantly associated with maintenance of salt and water homeostasis. It is probably controlled by interglomerular redistribution of blood flow and/or direct effects on renal tubular processing of salt and water. On the other hand, COX-1 appears to have a more defined role in the maintenance of glomerular filtration function. In general, treatment with celecoxib was associated with an incidence of peripheral edema greater than that noted with placebo and quantitatively comparable to that of conventional NSAIDs. The peripheral edema observed among celecoxib-treated subjects was not associated with significant weight gain (average increase less than 0.3 kg) or increased blood pressure. In the comparable data base which had been generated during the clinical development of rofecoxib, there was a tendency towards increasing degrees of peripheral edema with the use of increasing doses of this COX-2 specific inhibitor and the quantitative incidence of peripheral edema was similar to or greater than conventional NSAIDs.
4. Blood pressure
Renal PGs also acts to lower systemic vascular resistance. The generally modest blood pressure elevations of 3 to 6 mmHg observed during conventional NSAIDs therapy appears to be of little clinical consequence, but from an epidemiologic point of view, sustained increases as small as 5 to 6 mmHg in diastolic blood pressure over several years have been shown to result in a 67% increases in the risk of cardiovascular accident and a 15% increase in the risk of coronary artery disease (Fig. 6)25).
5. Hyperkalemia and renal tubular acidosis
Effects of prostaglandins on renal potassium transport have been described; NSAIDs-associated hyperkalemia appears to be primarily secondary to the effects on aldosterone rather than direct epithelial effects. Normal urine potassium excretion is dependent on two conditions; adequate sodium delivery to the collecting duct and the presence of aldosterone. Cyclooxygenase inhibition by NSAIDs results in two results; first, they decrease distal sodium delivery by reducing glomerular filtration and enhancing salt absorption by the nephron; second, by inhibiting macula densa PGs production they reduce renin release. By reducing renin release, angiotensin levels decrease, removing the major stimulus for adrenal aldosterone production. PG synthesis inhibition can lead to hyporeninemic hypoaldosteronism; plasma potassium rises by 0.2-0.6 mEq/dL. On the other hand, PGE2 was also reported to directly inhibit potassium secretion in the collecting duct26); thus loss of this inhibitory action would promote potassium secretion, mitigating rather than exacerbating hyperkalemia11). Serum potassium concentration begins to increase upon dosing and attains a new steady state within several days of onset of therapy18).
Hyperkalemia is a commonly reported and potentially dangerous complication of treatment with, especially, indomethacin which directly impairs cellular potassium uptake. Preexistent mild to moderate azotemia, potassium supplementation, potassium sparing diuretics, ACE inhibitor and/or angiotensin receptor blocker, diabetes, heart failure, old age are associated with an increased risk of the condition. NSAIDs induced hyporeninemic hypoaldosteronism may cause hyperkalemia/type 4 RTA in susceptible patients.
6. Acute renal failure
Non-selective NSAIDs do not significantly impair renal perfusion in normal subjects. However, NSAIDs can lead to reversible ischemia and renal insufficiency in disease states, particularly in hypovolemic disorders in which angiotensin II and norepinephrine secretion are increased. (Fig. 7). Typical features of NSAIDsinduced acute vasomotor renal failure include: the presence of one or more risk factors, modest salt and water retention with weight gain (1-2 kg), decreased urinary output, a low fractional sodium excretion (<1.0%), a benign urinary sediment, and prompt reversibility upon discontinuation of the NSAIDs.
Go to :
Traditional NSAIDs and selective COX-2 inhibitors
Selective COX-2 inhibitors also adversely affect renal function in such at risk patients who include those with volume depletion, heart failure, cirrhosis, intrinsic renal disease (eg, diabetic nephropathy), and hypercalcemia27). In a review of the literature, acute renal failure and/or severe electrolyte disturbances (particularly hyperkalemia and metabolic acidosis) were clearly precipitated by either celecoxib or rofecoxib. A study of 15 similar patients found that a single dose of high amounts of rofecoxib (250 mg) had significant and similarly detrimental effects on renal function as a single dose of indomethacin (75 mg)28). These observations suggest that selective COX-2 inhibitors should be avoided in patients with chronic renal insufficiency, severe heart disease, volume depletion, and/or hepatic failure.
Compared with celecoxib, the incidence of adverse events related to hypertension and edema were significantly higher with ibuprofen (but not diclofenac), and a decline in renal function was more apparent with diclofenac than with ibuprofen. These data provide a greater understanding of the relative effects of these agents on cardiorenal function and homeostasis29).
Go to :
Conclusion
Both COX-1 and COX-2 are constitutively expressed in the kidney. Prostaglandin enhances the renin secretion in the macula densa. PG induces natriuresis and diuresis by various mechanisms. Inhibition of renal prostaglandin synthesis by NSAIDs causes various electrolyte and acid-base disturbances including sodium retention (edema, hypertension), hyponatremia, and hyperkalemia/type IV RTA. NSAIDs associated electrolyte and acid-base disturbances are not uncommon in some clinical situations. Adverse renal effects of NSAIDs are generally associated with the PGs dependent states; volume-contracted states, low cardiac output, or other conditions that tend to compromise renal perfusion. Both non-selective and selective COX-2 inhibitors have similar renal adverse effects. In view of the many NSAIDs users' susceptibility to renal adverse effects due to their underlying disease or condition, physicians should be cautious in prescribing NSAIDs to susceptible patients.
Go to :
 XML Download
XML Download