

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
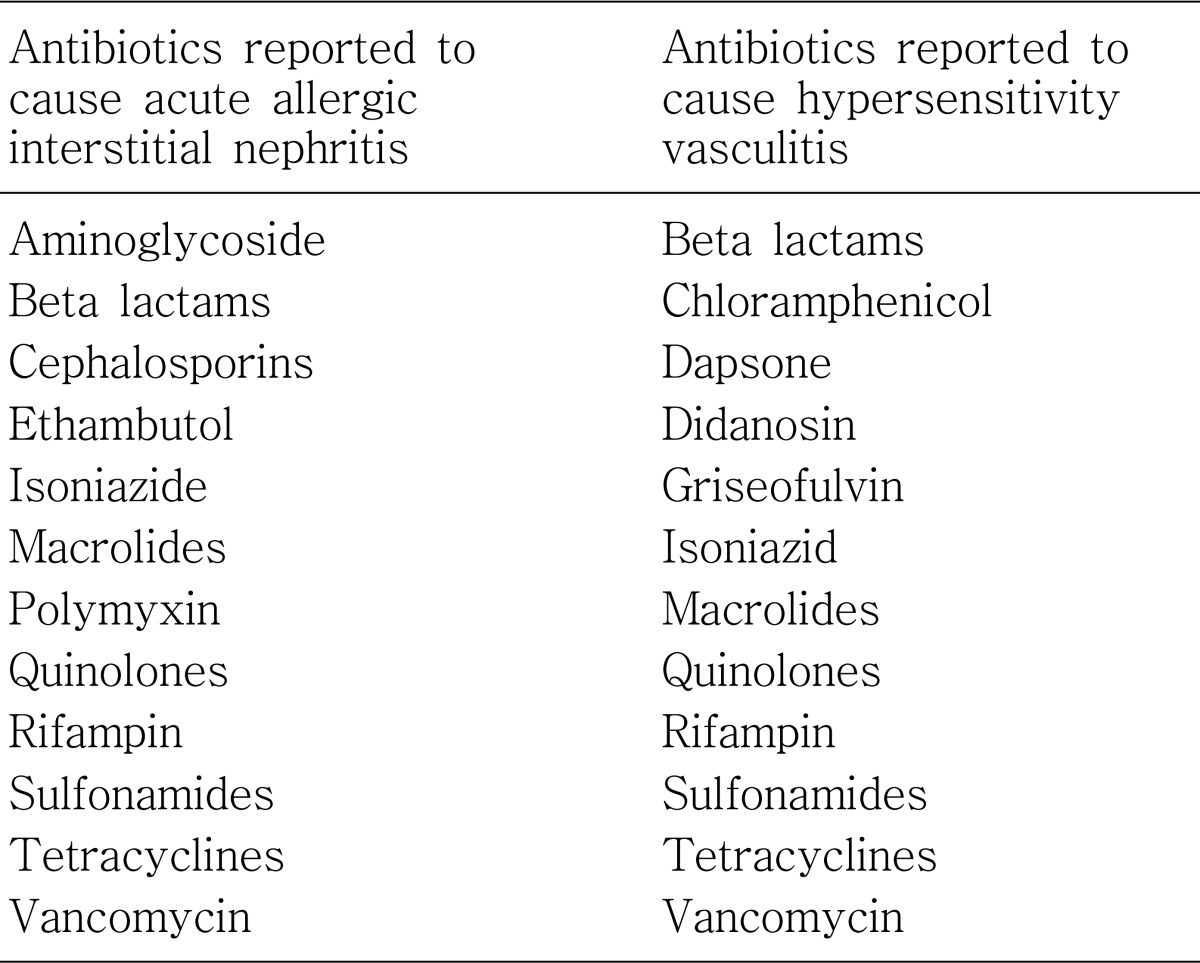
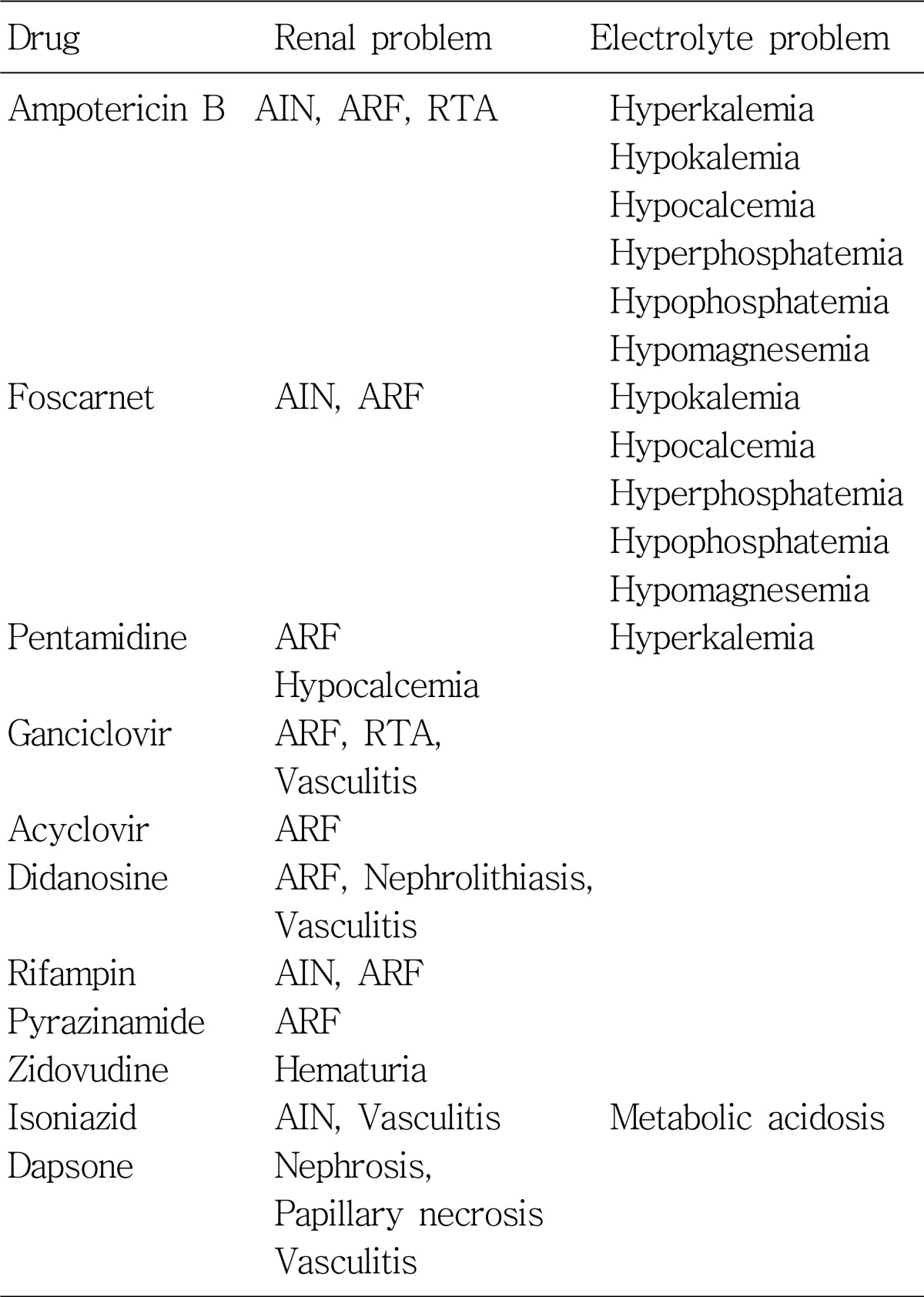
Adverse effects of antimicrobials are various and many of them are associated with development of both acute and chronic nephrotoxicity according to different mechanisms (Table 1). Sometimes, the electrolyte and acid/base disturbances can be developed without accompanying decrease in renal function, as in cases with aminoglycosides, amphotericin B, trimethoprim, and tetracycline. Table 2 shows the antimicrobials which may cause serious fluid and electrolyte imbalance.
Go to : 
Sodium and volume overload
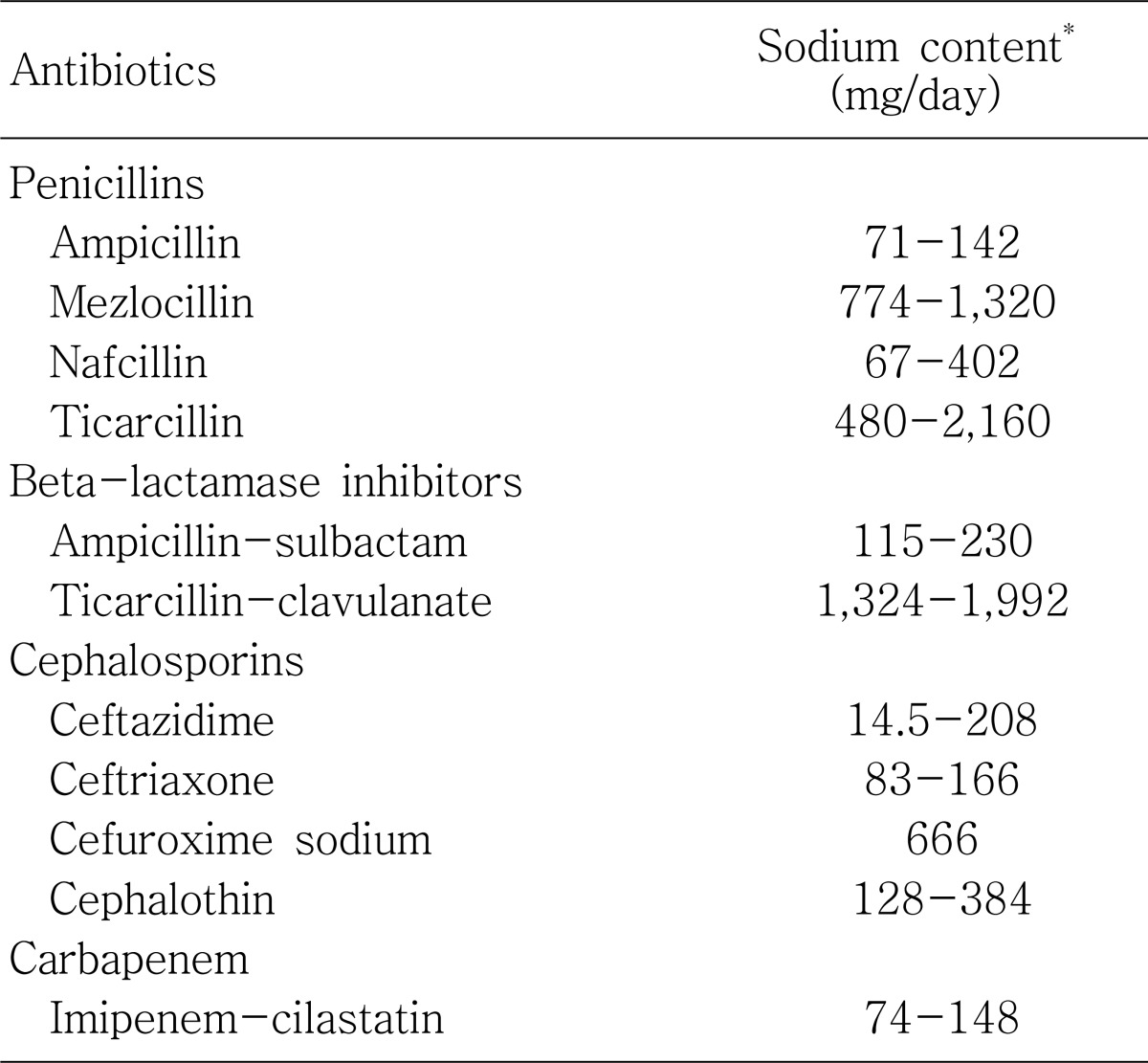
Certain antibiotics contain sodium or potassium salts, which may contribute to electrolyte disturbance. Because of the contained sodium, hypernatremia or fluid overload can develop. Thus, sodium-containing agents should be avoided in patients who are edematous or hypervolemic (such as elderly persons with congestive heart failure) (Table 3).
Go to :
Aminoglycoside
The aminoglycosides are the mainstay in the treatment of serious gram-negative systemic infections. They bind to the bacterial 30S ribosomal subunit, which translocates the peptidyl-tRNA from the A-site to the P-site and then inhibits the processing. The misreading of mRNA causes the bacterium to be unable to synthesize proteins vital to its growth. Aminoglycosides are primarily useful in infections involving aerobic, gram-negative bacteria; such as Pseudomonas, Acinetobacter, and Enterobacter. In addition, some Mycobacteria, including the bacteria that cause tuberculosis, are susceptible to aminoglycosides.
Aminoglycosides produce disturbances in electrolyte homeostasis, resulting in hypokalemia, hypomagnesemia, and hypocalcemia. The mechanisms of the aminoglycoside-induced syndome of hypokalemic metabolic alkalosis associated with hypomagnesemia are not clear. It has been suggested that animoglycosides may stimulate the renal tubular chloride channel, resulting in excessive urinary chloride loss1). The sodium chloride wasting in the renal tubule can lead to the stimulation of the renin-angiotensin-aldosterone axis, and subsequently hypokalemic metabolic alkalosis. Hypomagnesemia may be due to the coexistent hypokalemia. Recently, some reported data also suggest that as aminoglycosides are cationic, they act on extracelluclar polyvalent cation-sensitive receptors which are present in the distal convoluted tubular cells, and thus can inhibit hormone-stimulated magnesium absorption in this segment2). This activation of renal calcium-sensitive receptors would also lead to an increased calcium excretion. Interestingly, gentamicin administered at standard clinical doses causes immediate and transient renal calcium and magnesium wasting in normal humans3).
Aminoglycoside administration can cause a Fanconi-like syndrome consisting of renal glucosuria, phosphaturia, uricosuria, and metabolic hyperchloremic acidosis, which are the features of proximal tubular dysfunction1). Advanced age and long-term drug administration may predispose to renal tubular damage. Proximal tubular damage can be related to the toxic effects of these drugs. After aminoglycosides are filtered in glomeruli, some fractions (2-5%) are reabsorbed in the proximal tubules. These cationic particles electrostatically attach to anionic membrane phospholipids, resulting in lysosome swelling with phospholipid material and impaired generation of adenosine triphosphate. These effects inhibit energy formation, destroy normal cellular trafficking, disrupt apical membrane function, and culminate in cell dysfunction. Thus, the energy-depleted disordered cell metabolism or cell membrane damage can result in nephrotoxicity such as derangement in tubular function1).
Go to :
Ampbotericin B
Amphotericin B (AmB) is a polyene antifungal drug, often used intravenously for systemic fungal infections (e.g. in immunocompromised patients), like visceral leishmaniasis, Aspergillosis, cryptococcus infections (e.g. meningitis), and candidiasis. It was originally extracted from Streptomyces nodosus, a filamentous bacterium in 1955. Oral preparations of AmB are used to treat oral thrush; these are virtually nontoxic. As the mechanism of action, AmB associates with ergosterol, a membrane chemical of fungi and then forms a pore that leads to potassium leakage, resulting in fungal cell death.
AmB therapy can commonly cause hypokalemia and hypomagnesemia. It has been estimated that the incidence of hypokalemia secondary to AmB administration is as high as 75-90%. AmB-induced hypokalemia has been reported to be dose-dependent. Three separate mechanisms may be involved in the potassium wasting associated with AmB administration. First, AmB may induce intramembranous pore formation, or vacuolation, of the epithelial cells in the distal convoluted tubule of the kidney and in peripheral red blood cells by binding to cell membrane cholesterol. This mechanism is akin to its fungicidal action in which it binds to sterols in the fungal cell membrane to induce an efflux of cellular potassium. This action results in increased permeability of the distal convoluted tubule, with subsequent increased urinary potassium secretion through tubular Na+,K+-ATPase, and in an efflux of intracellular potassium from peripheral red blood cells4). The efflux of potassium from the red blood cells into the extracellular compartment produces a transient plasma hyperkalemia, with eventual renal excretion resulting in hypokalemia5). Second, AmB therapy may result in a defect in the distal tubule H+,K+-ATPase, or type 1 renal tubular acidosis (RTA), which increases the elimination of potassium. Type 1 RTA occurs with cumulative doses of AmB exceeding 2-3 g6). Finally, evidence exists that AmB affects ion transport in the distal colon in humans to enhance sodium reabsorption in the colonic epithelium, with resulting potassium excretion in the feces7).
The mechanism of AmB-induced hypomagnesemia is less clear. It has been hypothesized that AmB produces a defect that reduces magnesium reabsorption in the distal tubule8). The kidneys are not directly responsible for AmB-induced magnesium wasting. An alternative mechanism may be that the body distribution of magnesium is altered as a consequence of the cell membrane effects of AmB4). AmB can bind to the cholesterol moiety in human cells, causing intracellular magnesium leakage. This leakage may lead to transient increases in plasma magnesium concentrations, with eventual elimination through the kidneys, leading to hypomagnesemia.
Go to :
Trimethoprim/sulfamethoxazole
Trimethoprim/sulfamethoxazole (TMP/SMX) is a routinely prescribed antimicrobial used in the treatment and prophylaxis of various bacterial and protozoal infections. Use of TMP/SMX has been associated with the development of type 4 RTA and, more commonly, hyperkalemia. The mechanism by which TMP/SMX induces RTA is not well understood. Hyperkalemia itself may contribute to the development of acidosis through the effects on renal handling of ammonia and aldosterone secretion. The stimulation of aldosterone in this setting may attenuate the effects of TMP/SMX on handling of the hydrogen ion, therefore partially preventing acidosis9).
The mechanism by which TMP/SMX produces hyperkalemia is related to the trimethoprim component, which is a heterocyclic weak base and is structurally similar to the potassium-sparing diuretics like amiloride and triamterene10-12).
Hyponatremia was described in several cases with TMP/SMX. This may be related to the inhibitory effects of trimethoprim on sodium reabsorption in the cortical collecting tubules. Hyperchloremic non-anion gap metabolic acidosis was also reported in an AIDS patient treated for Pneumocystis carnii pneumonia with high-dose intravenous TMP/SMX (TMP 20 mg/kg/day)9).
Go to :
Tetracycline
Tetracyclines are a group of broad-spectrum antibiotics whose general usefulness has been reduced with the onset of bacterial resistance. It inhibits the action of prokaryotic 30S ribosome by binding aminoacyl-tRNA. Toxicity results from inactivation of mitochondrial 30S ribosomes in host cells.
The metabolic acidosis occurring with tetracycline antibiotics has mostly been associated with the use of outdated or degraded products. Tetracycline and its degradation metabolites accumulate within mitochondria of renal cells and interfere with oxidative phosphorylation. Metabolic acidosis secondary to proximal tubular dysfunction (type 2 RTA) may be present in isolation, or more commonly as part of the Fanconi syndrome, which consists of hypophosphatemia, hypouricemia, aminoaciduria, and glucosuria13).
Go to :
Miscellaneous antimicrobial agents
Renal tubular dysfunction has been reported with various other antimicrobial agents. The reverse transcriptase inhibitors (stavudine or lamivudine) are among those reported to cause RTA14). Mitochondrial dysfunction was postulated to be secondary to mitochondrial DNA depletion in renal tubular cells due to either stavudine or lamivudine.
Other antiretroviral agents like didanosine have been reported to cause proximal tubular dysfunction similarly to Fanconi syndrome15). Adefovir has also been reported to cause proximal tubular dysfunction16). Agents utilized in the treatment of cytomegalovirus infections have also been implicated in proximal tubular damage resulting in Fanconi syndrome, as reported with cidofovir17). Distal tubular damage with RTA has been seen with foscarnet18).
Go to :
Conclusion
Most of antimicrobials, especially vancomycin or aminoglycosides, are associated with acute renal failure caused by acute tubular necrosis, allergic interstitial nephritis, or rarely vasculitis. To avoid or prevent these serious complication associated with antimicrobials, early recognition and correction of their fluid-electrolyte disorders as well as other renal effects are warranted.
Go to :
 XML Download
XML Download