

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Hypernatremia, defined as a rise in the serum sodium concentration to a value exceeding 145 mmol per liter, is a common electrolyte disorder. Because sodium is a functionally impermeable solute, it contributes to tonicity and induces the movement of water across cell membranes. Therefore, hypernatremia invariably denotes hypertonic hyperosmolality and always causes cellular dehydration, at least transiently. Although correction of transient increases in plasma osmolality is usually well tolerated, correction of chronic plasma hypertonicity with rehydration therapy may be accompanied by brain swelling, herniation, and death1). The clinical differences between acute and chronic osmolar disorders can be understood through a consideration of the different mechanisms by which cells in the brain regulate their volume in response to brief and sustained osmotic challenges2).
Pathophysiology
As in other types of cells, hypertonically stressed brain cells regulate their volume initially by the rapid uptake of electrolytes3). With a prolonged elevation of plasma osmolality, however, most excess electrolytes in the brain are replaced by organic solutes. These solutes have historically been termed "idiogenic osmoles" because they were unidentified and thought to be produced by the brain cells themselves4). Studies in animals and cultured cells5) have revealed that idiogenic osmoles are the same organic osmolytes used by all organisms for volume regulation. The most important organic osmolytes in the mammalian brain include myo-inositol, taurine, glycerylphosphorylcholine, and betaine, which are accumulated primarily by uptake from extracellular fluids through the activation of sodium-dependent cotransporters5).
Beginning on the first day of the hypernatremia, brain volume is largely restored due noth to water movement from the cerebrospinal fluid into the brain (thereby increasing the interstitial volume)5) and to the uptake of solutes by the cells (thereby pulling water into the cells and restoring the cell volume)5-7). The latter response involves an initial uptake of sodium and potassium salts, followed by the later accumulation of osmolytes, which in animals consists primarily of myo-inositol and the amino acids glutamine and glutamate6, 7). Myo-inositol is taken up from the extracellular fluid via an increase in the number of sodium-myo-inositol cotransporters in the cell membrane8), whereas the source (uptake from the extracellular fluid or production within the cells) of glutamine and glutamate is at present unknown.
The cerebral adaptation in hypernatremia has two important clinical consequences: Chronic hypernatremia is much less likely to induce neurologic symptoms. Assessment of symptoms attributable to hypernatremia is often difficult because most affected adults have underlying neurologic disease. The latter is required to diminish the protective thirst mechanism that normally prevents the development of hypernatremia, even in patients with diabetes insipidus. Correction of chronic hypernatremia must occur slowly to prevent rapid fluid movement into the brain and cerebral edema, changes that can lead to seizures and coma9). Although the brain cells can rapidly lose potassium and sodium in response to this cell swelling, the loss of accumulated osmolytes occurs more slowly, a phenomenon that acts to hold water within the cells5). The loss of myoinositol, for example, requires both a reduction in synthesis of new sodium-inositol cotransporters8) and the activation of a specific inositol efflux mechanism in the cell membrane10). The delayed clearance of osmolytes from the cell can predispose to cerebral edema if the plasma sodium concentration is lowered too rapidly. As a result, the rate of correction in asymptomatic patients should not exceed 12 mEq/L per day, which represents an average of 0.5 mEq/L per hour.
Go to : 
Causes of hypernatremia
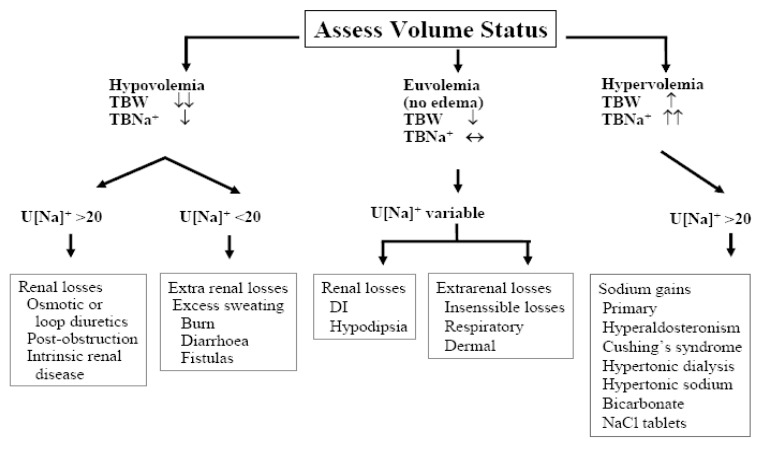
Hypernatremia represents a deficit of water in relation to the body's sodium stores, which can result from a net water loss or a hypertonic sodium gain (Table 1). Net water loss accounts for the majority of cases of hypernatremia1). It can occur in the absence of a sodium deficit (pure water loss) or in its presence (hypotonic fluid loss). Hypertonic sodium gain usually results from clinical interventions or accidental sodium loading. Because sustained hypernatremia can occur only when thirst or access to water is impaired, the groups at highest risk are patients with altered mental status, intubated patients, infants, and elderly persons11). Hypernatremia in infants usually results from diarrhea, whereas in elderly persons it is usually associated with infirmity or febrile illness12, 13). Thirst impairment also occurs in elderly patients14). Often the cause is evident from the history. Measurement of urine osmolality in relation to the plasma osmolality and the urine sodium concentration help if the cause is unclear (Fig. 1). Patients with diabetes insipidus present with polyuria and polydipsia (and not hypernatremia unless thirst sensation is impaired). Central diabetes insipidus and nephrogenic diabetes insipidus may be differentiated by the response to water deprivation (failure to concentrate urine) followed by the V2-receptor agonist desmopressin, causing concentration of urine in patients with central diabetes insipidus.

Go to :
Clinical manifestations
Signs and symptoms of hypernatremia largely reflect central nervous system dysfunction and are prominent when the increase in the serum sodium concentration is large or occurs rapidly (i.e., over a period of hours)15). Most outpatients with hypernatremia are either very young or very old16). Common symptoms in infants include hyperpnea, muscle weakness, restlessness, a characteristic high-pitched cry, insomnia, lethargy, and even coma. Convulsions are typically absent except in cases of inadvertent sodium loading or aggressive rehydration17, 18). Unlike infants, elderly patients generally have few symptoms until the serum sodium concentration exceeds 160 mmol per liter19). Intense thirst may be present initially, but it dissipates as the disorder progresses and is absent in patients with hypodipsia. The level of consciousness is correlated with the severity of the hypernatremia20). Muscle weakness, confusion, and coma are sometimes manifestations of coexisting disorders rather than of the hypernatremia itself (Table 2).

Brain shrinkage induced by hypernatremia can cause vascular rupture, with cerebral bleeding, subarachnoid hemorrhage, and permanent neurologic damage or death. Brain shrinkage is countered by an adaptive response that is initiated promptly and consists of solute gain by the brain that tends to restore lost water. This response leads to the normalization of brain volume and accounts for the milder symptoms of hypernatremia that develops slowly21). However, the normalization of brain volume does not correct hyperosmolality in the brain. In patients with prolonged hyperosmolality, aggressive treatment with hypotonic fluids may cause cerebral edema, which can lead to coma, convulsions, and death17, 18).
Go to :
Treatments
The primary goal in the treatment of patients with hypernatremia is the restoration of serum tonicity. In patients with hypernatremia that has developed over a period of hours, rapid correction of plasma sodium (falling by 1 mmol/L per hour) improves the prognosis without the risk of convulsions and cerebral edema1). Management of a shocked patient needs specialist input and close monitoring, preferably in a high dependency unit. Intravenous normal saline should be used to correct the extracellular fluid depletion, with calculation of the free water deficit to determine how much 5% dextrose to give. In patients with hypernatremia of longer or unknown duration, reducing the sodium concentration more slowly is prudent. Patients should be given intravenous 5% dextrose for acute hypernatremia or half-normal saline (0.45% sodium chloride) for chronic hypernatremia if unable to tolerate oral water. Central diabetes insipidus is treated with desmopressin, either as intranasal spray or tablets, with careful monitoring to avoid the complications of water intoxication (delaying one dose each week to allow polyuria and thirst to "breakthrough" in patients susceptible to hyponatremia with desmopressin may be prudent). Treatment of nephrogenic diabetes insipidus includes removal of precipitating drugs (if possible) and sometimes initiation of thiazide diuretics, non-steroidal anti-inflammatory drugs, or both. The following discussion primarily applies to the majority of patients in whom hypernatremia is induced by water loss.
1. Water deficit
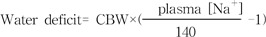
CBW refers to estimated current body water. The total body water is normally about 60 and 50 percent of lean body weight in younger men and women, respectively, and is somewhat lower in the elderly (about 50 and 45 percent in men and women, respectively)23). However, it is probably reasonable to use values about 10 percent lower (50 and 40 percent) in hypernatremic patients who are water-depleted. Thus, in a 60 kg woman with a plasma sodium concentration of 168 mEq/L, total body water is about 40 percent of body weight and the water deficit can be approximated from :
Water deficit=0.4×60 ([168/140]-1) = 4.8 liters
This formula estimates the amount of positive water balance required to return the plasma sodium concentration to 140 mEq/L. Then, when calculating the amount of free water to give (either intravenously, as dextrose in water, or orally if the patient is able to drink), insensible losses and some part of urine and gastrointestinal losses must be added to the calculation. Although this formula provides an adequate estimate of the water deficit in patients with hypernatremia caused by pure water loss, it underestimates the deficit in patients with hypotonic fluid loss.
2. Rate of correction
There are no definitive clinical trials, but data in children suggest that the maximum safe rate at which the plasma sodium concentration should be lowered is by 0.5 mEq/L per hour and no more than by 12 mEq/L per day24). Overly rapid correction is potentially dangerous in hypernatremia (as it is in hyponatremia). Hypernatremia initially causes fluid movement out of the brain and cerebral contraction that is primarily responsible for the associated symptoms. Within one to three days, however, brain volume is largely restored due to water movement from the cerebrospinal fluid into the brain and to the uptake of solutes by the cells24). Rapidly lowering the plasma sodium concentration once this adaptation has occurred causes osmotic water movement into the brain, increasing brain size above normal. This cerebral edema can then lead to seizures, permanent neurologic damage, or death17, 18). This sequence of an adverse response to therapy has been primarily described in children in whom the hypernatremia was corrected at a rate exceeding 0.7 mEq/L per h22). In comparison, no neurologic sequelae were induced if the plasma sodium concentration were lowered at 0.5 mEq/L per hour23).
3. Common errors in management
Isotonic saline is unsuitable for correcting hypernatremia. Consider a 50-year-old man with a serum sodium concentration of 162 mmol per liter and a body weight of 70 kg (estimated volume of total body water, 42 liters [0.5×70]). The retention of 1 liter of 0.9 percent sodium chloride will decrease the serum sodium concentration by only 0.22 mmol per liter ([154-162]÷[35+1]=-0.22). Although the sodium concentration of the infusate is lower than the patient's serum sodium concentration, it is not sufficiently low to alter the hypernatremia substantially. Furthermore, ongoing hypotonic fluid losses might outpace the administration of isotonic saline, aggravating the hypernatremia. The sole indication for administering isotonic saline to a patient with hypernatremia is a depletion of extracellular-fluid volume that is sufficient to cause substantial hemodynamic compromise. Even in this case, after a limited amount of isotonic saline has been administered to stabilize the patient's circulatory status, a hypotonic fluid (i.e., 0.2 percent or 0.45 percent sodium chloride) should be substituted in order to restore normal hemodynamic values while correcting the hypernatremia. If a hypotonic fluid is not substituted for isotonic saline, the extracellular-fluid volume may become seriously overloaded.
Extreme care must be taken to avoid excessively rapid correction or overcorrection of hypernatremia, which increases the risk of iatrogenic cerebral edema, with possibly catastrophic consequences. Selecting the most hypotonic infusate that is suitable for the particular type of hypernatremia ensures the administration of the least amount of fluid. Appropriate allowances for ongoing fluid losses must be made to prevent serious deviations in either direction from the targeted serum sodium concentration. Scrupulous adherence to these management guidelines should help prevent such complications. Most importantly, the fluid prescription should be reassessed at regular intervals in the light of laboratory values and the patient's clinical status1).
Go to :
Prognosis
The morbidity and mortality of patients with acute hypernatremia is very high. In children, the mortality of acute hypernatremia ranges between 10 and 70%. As many as two-thirds of the survivors have neurologic sequelae. In contrast, mortality in chronic hypernatremia is 10%. In adults, serum Na concentrations above 160 mmol/L are associated with a 75% mortality1). Since hypernatremia in adults occurs in the setting of serious disease the mortality rates may reflect the severity of the underlying diseases rather than that of hypernatremia.
Go to :
 XML Download
XML Download