

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Even though sodium transport occurs along the entire length of the tubule, the fine regulation in sodium reabsorption is primarily seen in the late distal tubule including the connecting tubule and collecting duct. This regulation is mainly mediated by local hormones such as aldosterone, vasopressin and angiotensin II which induced epithelial sodium channel (ENaC) activation1-8).
In the rat kidney, the three homologous subunits (α, β, and γ) that constitute the functional ENaC protein are detected in the late distal convoluted tubule (DCT2), connecting tubule (CNT), cortical collecting duct (CCD), and outer medullary collecting duct (OMCD), and to a lesser extent, the inner medullary collecting duct (IMCD)1-3). It has been shown that α-ENaC is mainly present at the apical domains of the principal cells, whereas β- and γ-ENaC are mainly associated with intracellular vesicles dispersed in the entire cytoplasm1). The physiological significance of the heterogeneity in the subcellular localization of the three sub-units has not been established. The α-subunit is functional when expressed alone in Xenopus laevis oocyte, but channel activity is highly increased by association with β- and γ-subunits4). The generation of gene knockouts of the individual subunits in mice demonstrated that altered expression of any of the three subunits has significant effects on multimeric ENaC protein sodium transport capacity5). Regulation of sodium reaborption by ENaC mediated by hormones such as aldosterone and vasopressin is associated with characteristic alterations in the expression of the individual ENaC subunits6, 7). Chronic aldosterone infusion in rats increases the protein abundance of α-ENaC. Moreover, aldosterone causes a mobility shift of γ-ENaC from an 85-kDa band to 70-kDa band8). Chronic vasopressin infusion results in significantly increased abundances of all three ENaC subunits6). The apical plasma membrane expression of ENaC can also be altered by changes in the trafficking of the channel subunits to the apical plasma membrane. Under sodium-replete conditions, when aldosterone levels are low ENaC immunostaining of CCD revealed diffuse labeling throughout the principal cells, consistent with localization in a vesicular pool. In contrast, sodium restriction or aldosterone infusion caused a dramatic redistribution of ENaC to the apical membrane7, 8). These results suggest that not only protein abundance but also translocation of ENaC to the apical plasma membrane are under the tight control.
The regulation of sodium transport in the distal nephron is paramount for the regulation volume and electrolyte balance in the body. In the collecting duct, electrogenic entry of sodium from the lumen into the cells is mediated by ENaC located in the apical plasma membrane9). This represents the rate-limiting step for sodium absorption and is characterized by inhibition with submicromolar concentrations of the diuretic amiloride10). Thus, ENaC is a critically important final regulator of the balance between intake and excretion of dietary sodium, and along with the thiazide-sensitive NaCl co-transporter constitutes the predominant sodium transport systems in the aldosterone-sensitive distal nephron.
Nephrotic syndrome and liver cirrhosis are mainly associated with avid sodium retention leading to the development of edema and ascites. However, the mechanism for the sodium retention is still incompletely understood and the molecular basis remains undefined. This review aims at providing recent insight into the role of ENaC and sodium (co)transporters in the kidney in sodium retentive disease such as nephrotic syndrome and liver cirrhosis.
Nephrotic syndrome
Puromycin aminonucleoside (PAN)-induced nephrotic syndrome is one of the most extensively studied models of glomerulonephritis in rats. This experimental model mimics the minimal change disease glomerulopathy found in human pathology. In vivo micropuncture studies in the unilateral model of PAN-induced nephrotic syndrome have shown that sodium reabsorption is specifically increased in the collecting duct and not in the proximal tubule and distal nephron11). Micropuncture of the accessible distal convoluted tubule revealed that the tubular sodium load was similar in PAN-treated kidney and control kidney of the same rat, and the final urine sodium excretion was threefold lower in urine collected from the PAN-treated kidney compared to that in the untreated control kidney in the same animal11). Thus, this study directly point to a role of increased sodium reabsorption in the collecting duct and, hence, it may be hypothesized that dysregulation of the key sodium channels and transporters in the collecting duct may be responsible for this. A few studies have subsequently demonstrated that sodium retention in PAN nephrotic rats is correlated with increased Na,K-ATPase activity and expression in the cortical collecting duct12, 13).
We demonstrated that PAN-induced nephrotic syndrome is associated with 1) sodium retention, decreased urinary excretion of sodium, a marked ascites, and increased plasma aldosterone level, 2) upregulation of protein abundance of specific ENaC subunits in cortex, outer medulla and inner medulla, and 3) increased apical targeting of ENaC subunits in connecting tubule and collecting duct segments observed as increased immunolabeling in apical plasma membrane domains14). In contrast, the protein levels of other major sodium transporters expressed in nephron segments at a site proximal to the connecting tubule (i.e., NHE3, NKCC2, Na,K-ATPase and NCC) were significantly reduced in these segments14). Indeed, there was no downregulation of the Na,K-ATPase expression in the collecting duct. Taken together, these observations therefore strongly support the view that the renal sodium retention associated with PAN-induced nephrotic syndrome is caused by increased sodium reabsorption in connecting tubule and collecting duct14). Thus, this is likely to represent a key molecular basis for the sodium retention associated with PAN-induced nephrotic syndrome combined with the previously demonstrated increase in collecting duct Na,KATPase activity and protein abundance12, 13). Moreover, our results extend this study by demonstrating the segmental specific upregulation of all three ENaC subunits, and most importantly provide evidence for a significantly increased apical targeting by immunoelectron microscopy. The latter is essential since it is important to document that the increased expression is at the site of function i.e. in the plasma membrane. Finally, we also confirm that there is a uniform downregulation of all investigated sodium transporters in the thick ascending limb of Henle (TAL) and proximal tubule. This together with observed increased expression and increased targeting of ENaC subunits in combination with the previous functional micropuncture studies strongly support the view that the sodium retention in the nephrotic syndrome may occur in the connecting tubule and collecting duct15).
Nephrotic syndrome may develop as a result of primary diseases such as minimal change disease or immune glomerulonephritis. Membranous glomerulonephritis (MGN) remains the most common cause of primary nephrotic syndrome in adult. A further reason for its importance is that approximately 25 to 50% of patients progress to end-stage kidney disease over 10 years. Thus, MGN has a different and more progressive clinical course as compared to the non-progressive benign character of minimal change nephrotic syndrome16). Mercury chloride has been known to induce a systemic autoimmune disease including membranous nephropathy with IgG deposits. This nephropathy is responsible for the development of high-range proteinuria and a full-blown nephrotic syndrome associated with generalized edema and ascites17). To elucidate whether the changes in expression and plasma membrane targeting of ENaC subunits in the PAN-induced minimal change nephrotic syndrome (caused by podocyte injury, not immune complex mediated) is unique to the PAN model or may be a more general characteristic of nephrotic syndrome including immune complex mediated glomerulonephritis, and to investigate the physiologic role of changes in subcellular distribution of ENaC subunits in the abnormal sodium retention and ascites formation during disease states, we examined the changes of ENaC abundance and/or trafficking and 11HSD2 in HgCl2 induced nephritic syndrome in Brown Norway rats. The results demonstrated that HgCl2-induced immune complex glomerulonephritis was associated with 1) sodium retention, decreased urinary sodium excretion, development of ascites, and increased plasma aldosterone level; 2) increased abundance of αENaC (Fig. 1) 3) increased apical targeting of ENaC subunits in connecting tubule and collecting duct segments; and 4) decreased protein abundance of 11βHSD2 (Fig. 2)18). These observations therefore strongly support the view that the renal sodium retention associated with HgCl2 induced nephrotic syndrome is caused by increased sodium reabsorption in the distal nephron including the connecting tubule and collecting duct. These results are consistent with the results of our previous studies of nephrotic syndrome induced by PAN14). Thus, a straightforward interpretation of these observations would be that increased apical targeting of ENaC subunits plays a role in the development of sodium retention in HgCl2 nephrotic syndrome as well, and it could be argued that this mechanism in fact could be a general characteristic of the nephrotic syndrome including both minimal change disease and immune complex mediated glomerulonephritis.
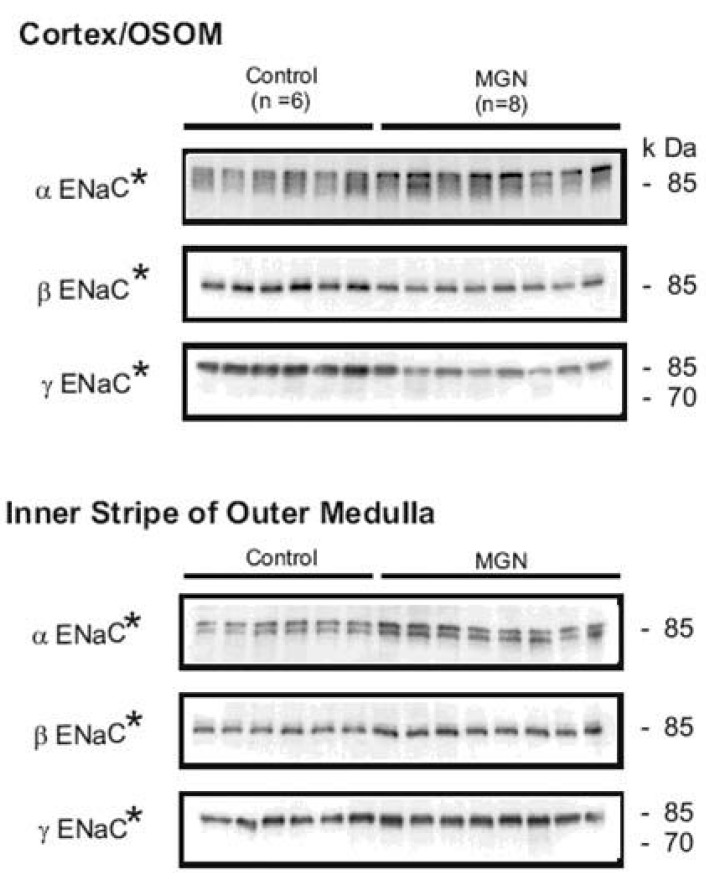 | Fig. 1Semi-quantitative immunoblots of kidney protein prepared from cortex/outer stripe of outer medulla (OSOM) and inner stripe of outer medulla (ISOM) from control and HgCl2-nephropathic rats (membranous glomerulonephritis, MGN). The protein abundance of αENaC was increased in the cortex/OSOM and ISOM. The protein abundances of βENaC and γENaC were decreased in the cortex/OSOM, while increased in the ISOM. *p<0.05 vs. control. (Kim et al., Am J Physiol Renal Physiol 2006).
|
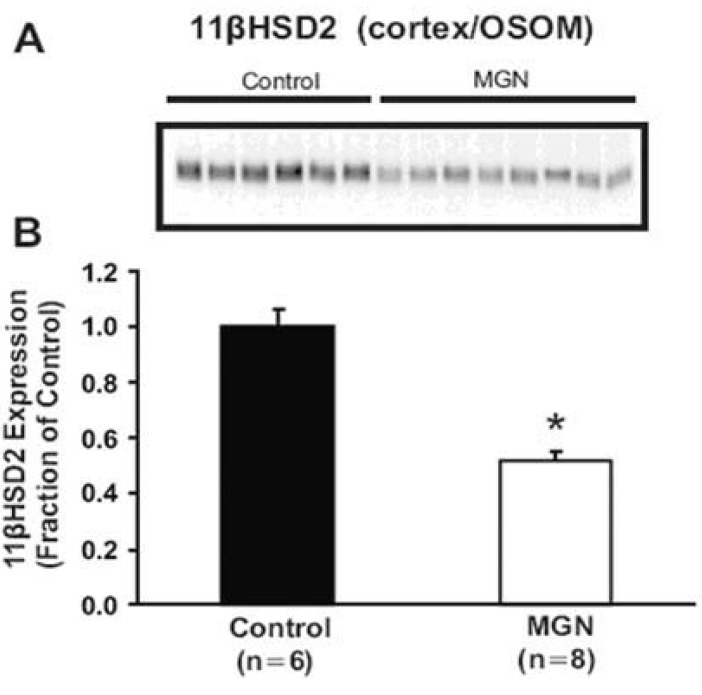 | Fig. 2Semiquantitative immunoblotting and immunoperoxidase microscopy of 11βHSD2 from control and HgCl2-nephropathic rats. A) Immunoblot was reacted with anti-type 2 isoform of 11β-hydroxysteroid dehydrogenase (11βHSD2) antibodies and revealed a single 44 kDa bands. B) densitometric analyses revealed that 11βHSD2 expression in the cortex/OSOM was significantly reduced in HgCl2-nephropathic rats compared with controls. *p< 0.05 vs. control (Kim et al., Am J Physiol Renal Physiol 2006).
|
Apical targeting of ENaC subunits and protein abundance of αENaC can be regulated by aldosterone7,8). Indeed, plasma aldosterone levels were significantly increased in HgCl2 nephropathy, and thus, it can be speculated that aldosterone stimulate the sodium reabsorption in HgCl2 nephropathy. However, substantial evidence argue against a major role of aldosterone in nephrotic syndrome : 1) ascites and edema occur in rats with nephrosis attributable to rabbit anti-rat kidney serum injection, without elevated aldosterone levels19). 2) inhibition of angiotensin converting enzyme by captopril failed to induce natriuresis in rats with PAN nephrosis despite decreased aldosterone levels20). and 3) sodium retention was observed only in the affected proteinuria kidney in rats unilateral models of PAN nephrosis11). These observations suggest that aldosterone may not alone be involved in the sodium retention and presumably in the ENaC regulation in the nephrotic syndrome.
It has been suggested that the activity of type 2 11βhydroxysteroid dehydrogenase (11βHSD2) can regulate the sodium reabsorption in the aldosterone responsive renal tubules by glucocorticoid induced activation of mineralocorticoid receptor (MR)21). We also demonstrate that there is a downregulation of 11βHSD2 protein expression in the kidney in HgCl2 nephrotic syndrome as well as in PAN nephrosis. This finding is in line with recent studies in nephrotic patients22) and in experimental nephrotic syndrome induced by PAN or adriamycin23). The investigators observed a rise in the plasma ratio of corticosterone/11-dehydrocorticosterone as well as in the urinary ratio of (THB+5α-THB)/THA in nephritic rats23), and interpreted this change as a result of a decreased activity of 11β-hydroxysteroid dehydrogenase. We here propose that this may occur via a downregulated protein expression of 11βHSD2 in nephrotic syndrome. These findings suggest that downregulation of 11βHSD2 provides access of glucocorticoids to the MR, resulting in increased activity of MR and collecting duct sodium retention in rats with nephrotic syndrome.
Systemic and local factors are known to regulate ENaC abundance, targeting and activity as well as Na,K-ATPase activity. These include aldosterone7, 8), vasopressin6) and angiotensin II (ANG II)24). Aldosterone is known to increase Na,K-ATPase activity in the cortical collecting duct and to increase ENaC redistribution to the apical membrane of connecting tubule and collecting duct principal cells and to promote the expression of a 70 kDa form of the gamma ENaC subunit, thought to be linked to activation of the channel by proteolytic cleavage7, 8). It is therefore tempting to link this hormone with the findings observed in PAN nephrosis. Moreover levels of vasopressin, ANG II and aldosterone are increased in the initial phase of nephrosis14, 25, 26), leading to the hypothesis that they may play significant roles in the development of sodium retention. As the time course and magnitude of sodium retention is indistinguishable in nephrotic Brattleboro rats (vasopressin deficient) compared to normal nephrotic rats12), vasopressin is not considered to be a major factor for the development of sodium retention in nephrotic syndrome. We have also demonstrated that ANG II AT1 receptor blockade, candesartan, did not improve the sodium retention associated with PAN-induced nephrotic syndrome nor the time course of its development27). The situation is less clear for aldosterone. There have been contradictory studies on the impact of aldosterone absence on the development of edema in nephrotic syndrome28, 29). Furthermore, even though pharmacologic blockade of aldosterone has been proven to be unsuccessful regarding sodium retention in experimental nephrosis15), this cannot rule out aldosterone effect as pharmacologic mineralocorticoid receptor blockade has been demonstrated to be useless against ENaC trafficking30) and would not avoid aldosterone action through the glucocorticoid receptor31). Finally, Na,K-ATPase in the collecting duct of nephrotic rats is still activated in the absence of aldosterone29). However nothing is known about ENaC regulation in that condition. It is therefore not known whether the enhanced ENaC targeting observed in PAN nephrosis is a primary dysregulation in nephrotic syndrome or is secondary to the high aldosterone levels observed in this condition.
We therefore studied the development of nephrotic syndrome in adrenalectomized rats, aiming at defining the role of aldosterone on sodium retention and ENaC targeting observed in this pathologic condition. Puromycin treatment of adrenalectomized rats supplemented with dexamethasone induced sodium retention in spite of the absence of aldosterone. Immunocytochemical analyses revealed absence of enhanced apical targeting of ENaC subunits in PAN treated adrenalectomized (ADX-PAN) rats with distribution of labelling similar to adrenalectomized dexamethasone-treated control rats (ADX). Moreover ENaC subunit abundance was increased in ADX-PAN rats32). Key sodium transporters were downregulated as previously observed in non-adrenalectomized puromycin-treated rats, whereas the global expression of the alpha-1 subunit of the Na,K-ATPase was unchanged. Taken together, PAN treatment in the absence of aldosterone induced sodium retention, increased ENaC expression but did not change the subcellular distribution of ENaC. This indicates that the previously observed enhanced apical targeting of ENaC in PAN induced neprotic syndrome is caused by aldosterone and that development of sodium retention can occur in the absence of aldosterone in nephrotic syndrome. However, it is not entirely clear whether whether puromycin-treatment results in an increased sensitivity of kidneys to systemic hormonal changes and which local factors might influence the sodium retention and ENaC activity.
Go to : 
Liver cirrhosis
Renal sodium and water retention has been shown to be responsible for the development of ascites not only in patients with liver cirrhosis but also in experimental cirrhotic rats. Atrial natriuretic peptide resistance, arginine vasopressin (AVP), renin-angiotensin-aldosterone, and sympathetic nerve overactivity have been shown to modulate renal tubular functions and have been considered to be mainly involved in sodium and water retention in liver cirrhosis. In kidneys of cirrhotic rat, increased reabsorption of sodium and water in the distal nephron and collecting duct has been suggested to be one of the most important renal tubular dysfuctions involved in the pathogenesis of ascites. However, the underlying molecular and cellular mechanisms for the sodium retention are still incompletely understood. In particular, the role of aldosterone in sodium retention and ascites formation in liver cirrhosis is still unclear. Previous studies demonstrated that plasma aldosterone levels were usually elevated in liver cirrhosis with ascites33) and that spironolactone, a mineralocorticoid receptor antagonist, increased sodium excretion in these patients34). This suggests that hyperaldosteronism is of major importance in the pathogenesis of sodium retention.
There has been some controversy as to the role of regulation of the renal sodium transporters and channels in the development of edema in animal models of cirrhosis. There are two major rat models of liver cirrhosis. In the first, rats are treated with carbon tetrachloride (CCl4) in combination with Phenobarbital supplied in the drinking water, which results in liver toxicity. In the second, rats have their common bile duct ligated, and increased circulating levels of bile acids (cholestasis) results in hepatotoxicity and eventually cirrhosis. Differences in the severity, reproducibility, and rapidity of the manifestation of ascites in the two models probably play a role in some discrepancies35).
1. CCl4 induced liver cirrhosis
The renal responses for sodium retention displayed wide variations among the rats with CCl4-induced liver cirrhosis. Some rats showed markedly decreased urinary sodium excretion (sodium retaining stage, group A) whereas the others exhibited unchanged urinary sodium excretion (maintenance stage, group B) compared with controls, even though all CCl4-treated rats had significant amount of ascites (Fig. 3)36). The results demonstrated that CCl4-induced sodium retaining stage (group A) of liver cirrhosis was associated with 1) decreased urinary sodium excretion, and increased or maintained plasma aldosterone levels; 2) increased apical targeting of ENaC subunits in DCT2, CNT and collecting duct segments; and 3) decreased protein abundance of 11βHSD2. In contrast, maintenance stage (group B) of liver cirrhosis was associated with no changes in the urinary sodium excretion, trafficking and abundance of ENaC subunits and the abundance of 11βHSD236). The most important finding is the striking increase in targeting of all ENaC subunits to the apical plasma membrane domain in DCT2, CNT and collecting duct in the sodium retaining stage (group A), but not in the maintenance stage (group B) of liver cirrhosis. Since an increased targeting of all ENaC subunits to the apical plasma membrane is associated with increased sodium reabsorption, the present finding of an increased ENaC targeting in the sodium-retaining stage of liver cirrhosis could contribute significantly to the increased renal tubular sodium reabsorption. These observations therefore strongly support the view that the renal sodium retention in the decompensated liver cirrhosis is mainly caused by an increased sodium reabsorption in distal nephron including the collecting duct37).
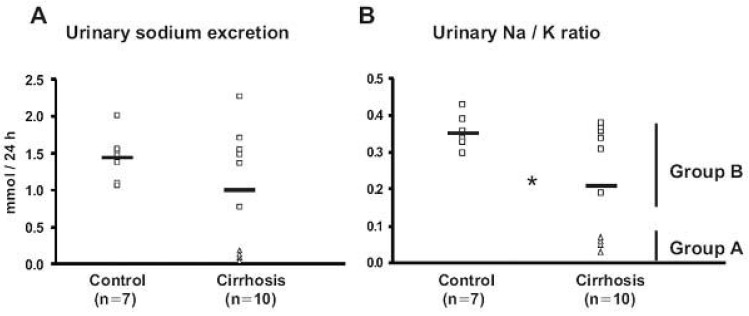 | Fig. 3Urinary sodium excretion (A) and Urinary Na/K ratio (B) from control (Control) and CCl4-treated cirrhotic rats (Cirrhosis) in protocol 1. A) Urinary sodium excretion was similar in control and CCl4-induced cirrhosis rats. Four (△, Group A) of the CCl4-induced cirrhosis rats showed markedly decreased urinary sodium excretion, while the other six rats (□, Group B) remained similar compared with controls. B) The urinary Na/K ratio was decreased in cirrhotic rats, indicating increased aldosterone effectiveness in the distal nephron. *p<0.05 vs. control (Kim et al., J Am Soc Nephrol 2005).
|
We demonstrated that the abundance of βENaC was decreased or unchanged, and the abundance of the 70-kDa form of γENaC was increased while the 85-kDa band was markedly decreased in the sodium-retaining stage (group A) of cirrhotic rats (Fig. 4)36). Previous studies demonstrated that aldosterone causes a mobility shift of γENaC from an 85 kDa band to 70 kDa band without a change in total γENaC protein abundance7). The same changes are observed in chronically sodium-restricted rats in addition to a significant downregulation of the βENaC subunit7). Thus, the observed increased apical targeting and altered expression of β- and γENaC subunits in group A could be caused by the stimulation of mineralocorticoid receptor (MR) in the aldosterone-responsive epithelium.
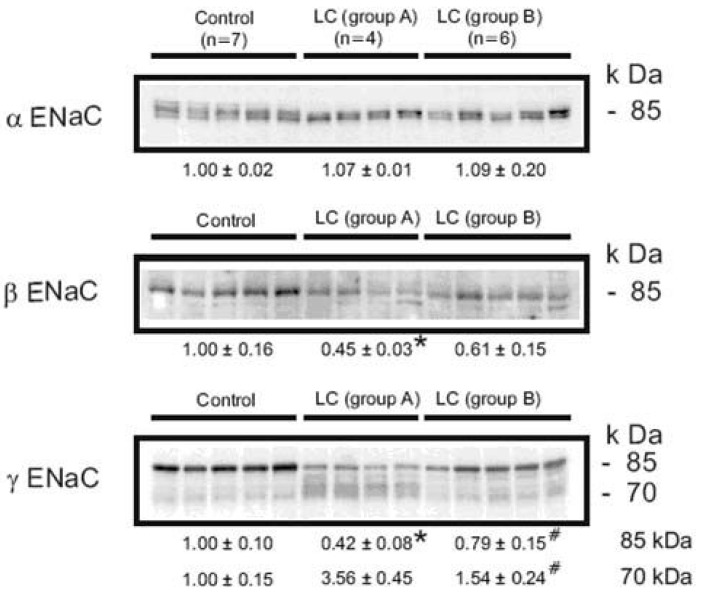 | Fig. 4Semi-quantitative immunoblots of kidney protein prepared from cortex/OSOM of control and CCl4-treated cirrhosis rats subdivided into group A or group B liver cirrhosis in protocol 1. In group A, protein abundance of αENaC was unchanged, while βENaC abundance was decreased compared with controls. The γENaC underwent a complex change associated with the increased abundance of 70 kDa band with a concomitant decrease in the main 85 kDa band. In contrast, there were no significant changes of ENaC subunit expression in group 2. *p<0.05 vs. control, #p<0.05 vs. group A (Kim et al., J Am Soc Nephrol 2005).
|
2. Common bile duct ligation (CBDL) induced-biliary liver cirrhosis
Long-term bile duct ligation in rats is a well known experimental model of liver cirrhosis and portal hypertension, and has been widely used to study the renal mechanisms of sodium retention, as well as systemic and splanchnic hemodynamics38). CBDL rats showed decreased fractional excretion of sodium and hence, positive sodium balance in the sodium retaining stage of liver cirrhosis (at 6 wks). It is suggested that increased renal tubular absorption of sodium may account for the sodium retention and pathogenesis of ascites formation. It is noteworthy that at 8 weeks of liver cirrhosis we did not see any evidence of positive sodium balance even though all rats showed marked ascites39). These findings may suggest that there are dynamic changes in sodium retention at different stages (i.e., a sodium retaining stage and a compensatory stage) of liver cirrhosis. It should be emphasized that the two time periods are two in a continuum of changes during the development of sodium retention and the stages with partial of full compensation. Recognition of this apparent biphasic renal response to cirrhosis, with progression from the early to the late phase of decompensation, may help to clarify apparent discrepancies in a variety of earlier studies.
The most important finding is the increased targeting of all ENaC subunits to the apical plasma membrane domain in DCT2, CNT and collecting duct in the sodium retaining cirrhotic rats (Fig. 5), but not in 8 week liver cirrhotic rats without positive sodium balance39). Since increased ENaC plasma membrane targeting is strongly associated with increased sodium reabsorption, this finding very strongly suggests that increased ENaC targetings contribute significantly to the increased renal tubular sodium reabsorption. The increased apical targeting of ENaC subunits in CBDL-induced liver cirrhosis may be caused by increased aldosterone activity via stimulation of mineralocorticoid receptor (MR) in the aldosterone responsive epithelium. Since there was a markedly enhanced apical plasma membrane expression of all ENaC subunits at 6 wk liver cirrhosis associated with maintained plasma aldosterone levels but not at 8 wk liver cirrhosis where aldosterone levels were markedly reduced, it is likely that aldosterone plays an essential role. However, other mechanisms may also contribute as discussed below.
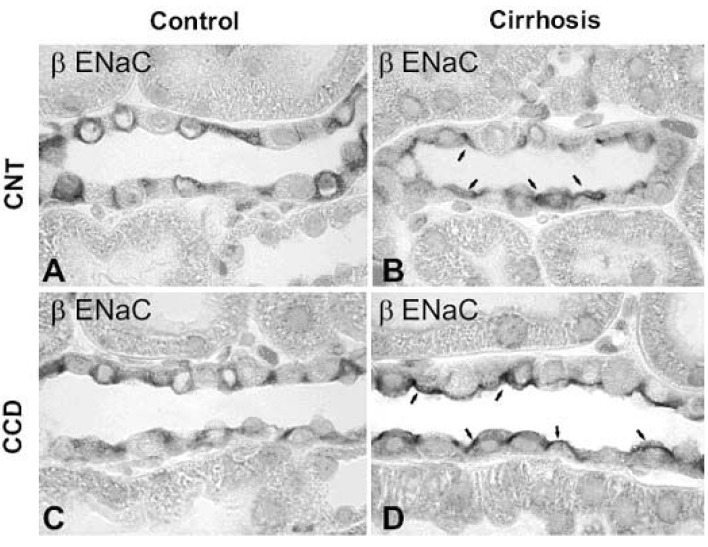 | Fig. 5Immunoperoxidase microscopy of βENaC in the connecting tubule (CNT) and cortical collecting duct (CCD) from control and 6 wk CBDL-induced liver cirrhosis rats. Immunoperoxidase labeling of βENaC is mainly associated with the entire cytoplasm of principal cells of the CNT (A) and CCD (C) in control rats. In contrast, βENaC labeling was markedly redistributed to the apical plasma membrane domains in CNT (B) and CCD (D) in 6wk cirrhotic rats. Arrows indicate apical labeling. Magnification : ×630. (Kim et al., Kidney Int 2006)
|
In vitro studies have demonstrated that the MR has an equal affinity for aldosterone and glucocorticoids, yet in vivo it displays specificity for aldosterone in the face of much higher circulating concentrations of glucocorticoids. This specificity is conferred by 11βHSD2, an enzyme that converts glucocorticoids to inactive 11-ketosteroid derivatives that have weak or no affinity for the MR in mineralocorticoid sensitive tissues including the distal nephron21). Loss of function mutation of 11βHSD2 or inhibition of 11βHSD2 activity allows glucocortiocids to promote renal sodium retention and potassium excretion in the cortical collecting tubule40, 41). These findings suggest that the 11βHSD2 regulates the sodium reabsorption in the aldosterone responsive renal tubules by glucocorticoid-induced activation of the MR. We demonstrate in the present studies that there is downregulation of 11βHSD2 abundance in the kidney in decompensated liver cirrhosis rats. This finding is in line with recent studies in compensated liver cirrhosis reported by Quattropani et al.42) and Escher et al.43). They demonstrated that 11β HSD2 activity in the kidney was reduced in liver cirrhosis following bile duct ligation42, 43). These findings together suggest that reduced expression and activity provides basis for access of glucocorticoids to the MR, thereby promoting increased collecting duct sodium reabsorption despite normal plasma levels of aldosterone in rats with experimental liver cirrhosis. This notion was strongly supported by the increased apical targeting of ENaC subunits in rats with glycyrrhizic acid-induced inhibition of 11βHSD2 activity44).
Plasma aldosterone levels, at least in humans and rats without liver cirrhosis, have been shown to be reduced in conditions with low expression of 11βHSD240, 41). It is noteworthy that plasma aldosterone levels were not reduced in 6 wk liver cirrhosis rats, despite that the protein abundance of 11βHSD2 was significantly decreased. Thus, coordinated activation of MR by both glucocorticoid as a consequence of reduced activity of 11βHSD2 and by relatively high plasma aldosterone may enhance distal renal tubular sodium reabsorption and potassium loss, and hence play important roles for the pathogenesis of sodium retention and possible ascites formation in CBDL-induced liver cirrhosis. In contrast, in response to liver cirrhosis for 8 weeks ENaC abundance was decreased (Fig. 6) and the reduction was associated with decreased plasma aldosterone levels39). In addition, the increased apical targeting of ENaC subunits was retrieved in spite of decreased abundance of 11βHSD2. It can be speculated that 11βHSD2 is still functioning but not at full power (thus aldosterone may play a major role), or it may be speculated that differences in ENaC trafficking between 6 and 8 weeks are independent of aldosterone and/or 11βHSD2.
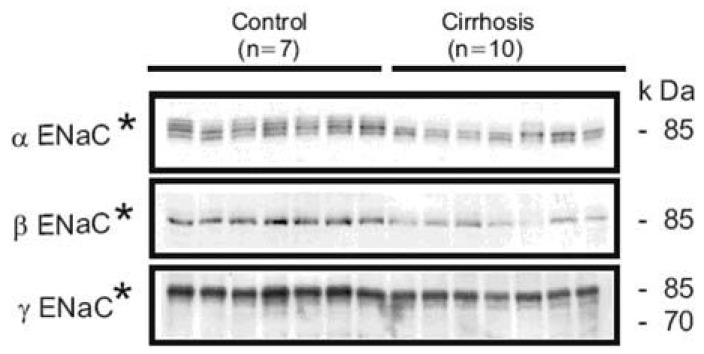 | Fig. 6Semiquantitative immunoblotting of kidney protein prepared from cortex/OSOM from control and 8 wk CBDL-induced liver cirrhosis rats. The protein abundance of all ENaC subunits (αENaC, βENaC and γ ENaC) was markedly decreased in cortex/OSOM in 8 wk liver cirrhosis rats compared with controls. *p<0.05 vs. control (Kim et al., Kidney Int 2006).
|
In normal rats both the apical targeting and protein abundance of αENaC are increased by high dose aldosterone treatment7). However, in the present study we did not observe an increase in αENaC protein expression in the cortex in kidneys from rats with CBDL-induced liver cirrhosis for 6 weeks although increased abundance of αENaC was seen in inner stripe of the outer medulla. It may be speculated that glucocorticoids may be involved in this regulation, and if this is the case the observations suggest that regulation of αENaC abundance is not as sensitive, as is the regulation of changes in the subcellular localization of ENaC, to MR activation by glucocorticoids. Whether the differential regulation of ENaC subunit targeting and expression may be related to differences in the kinetics of MR-ligand interaction remains unknown; aldosterone dissociates more slowly from MR than cortisol does, indicating that the stability of the aldosterone-MR complex is higher than that of the cortisol-MR complex45, 46). In addition, the MR activity can be regulated by at the level of transcription activation or repression by cell specific co-factors47).
It is also interesting to note that the rats with liver cirrhosis apparently exhibited dynamic changes in sodium retention and ENaC trafficking. After 6 weeks, liver cirrhosis animals exhibited sodium retention, potassium loss and decreased urine Na/K ratio indicating increased aldosterone-like activity. These findings can be explained in part by decreased abundance of 11βHSD2, which permits the stimulation of MR by glucocorticoid resulting in increased apical targeting of ENaC subunits. On the other hand, at 8 weeks the rats with liver cirrhosis did not exhibit any changes in urinary sodium and potassium excretion even though they all had marked ascites. The underlying mechanism for the time course changes in circulating aldosterone levels and changes in ENaC subunits trafficking remains undefined. However, it can be speculated that at later stages of liver cirrhosis the rise in extracellular fluid volume and enhanced secretion of atrial natriuretic peptaide (ANP) contribute to the decrease in plasma aldosterone levels and, hence, limit ENaC abundance/trafficking since ANP has been known to directly inhibit aldosterone secretion in the adrenal glands through specific ANP receptors in the zona glomerulosa48).
The results strongly suggests that the marked increase in apical ENaC subunit targeting combined with diminished abundance of 11βHSD2 in the DCT2, connecting tubule and collecting duct is likely to play key roles for the sodium retention associated with CBDL-induced liver cirrhosis in rats in the sodium retaining stage (i.e. in this model after 6 weeks of CBDL). The subsequent decrease in the expression of ENaC subunits and the reduced targeting after 8 weeks of CBDL-induced liver cirrhosis may contribute to promote sodium excretion at later stages of liver cirrhosis. This pahse may represent active and successful escape from the sodium retention by means of down-regulation of the activity of the ENaC.
Go to :
 XML Download
XML Download