

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Liver tumors of mesenchymal origin are rare and account for approximately 1.0% of all primary hepatic tumors.1 Angiosarcomas, leiomyosarcomas, fibrosarcomas, embryonic undifferentiated sarcomas and unspecified sarcomas are the most common types of primary mesenchymal tumors of the liver.2 Primary hepatic leiomyosarcoma are rare tumors of the liver, and usually arise from intrahepatic vascular structures, bile ducts or ligamentum teres.345
Presentations of the tumor usually are non-specific and diagnosis often is delayed until the tumor reaches a large size. Consequently, this has a negative impact on the prognosis regardless of therapeutic management. Due to the rare appearance of those tumors it is difficult to establish a standard of care and specific recommendations. Surgery combined with chemotherapy is the standard of care. We herein report a case of primary hepatic leiomyosarcoma, treated surgically.
CASE
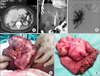
A 69-year-old female with a large hepatic mass had been admitted previously in another hospital where she underwent an operation. The proceeding was an open-close and the tumor was characterized as inoperable. The patient was referred to our department for a second opinion. Her chief complaint was pain in the abdomen and a palpable abdominal mass. She had no history of liver disease hepatitis or alcohol abuse. Her past medical history and family history were unremarkable. Physical examination revealed a marked hepatomegaly extending from below the right costal margin to the stomach. Laboratory analysis revealed normal liver function tests. Serum albumin level and prothrombin time were within normal range. White blood cell count, platelet, α-fetoprotein (AFP), CA 19-9 and Carcinoembryonic antigen (CEA) were normal. The patient had negative hepatitis C virus antibody and hepatitis B surface antigen (HBsAg). Abdominal computed tomography (CT) revealed a hypodense lesion (14 cm×11 cm) on plain scans, heterogeneous enhancing lesion on arterial phase and delayed washout on portal venous phase occupying segments IVA, V, VI and part of VII (Figs. 1A and B). Chest CT, upper gastrointestinal and lower gastrointestinal endoscopy revealed no significant pathology. From the previous operation the pathology report diagnosed the tumor as lipomyosarcoma. Due to the extent of the tumor and the location, portal vein embolization (PVE) was conducted. PVE of the right portal vein and selective empolization of the branch of segment IV was conducted (Fig. 1C). Five weeks later a new CT scan revealed increase in FLR and surgical exploration was undertaken. A huge tumor measured 14 cm×11 cm prolapsing from the liver infiltrating the greater omentum adjacent to the transverse colon was revealed (Fig. 1D). En-block resection of the omentum and the abdominal wall was conducted. Additionally, resection of segments IVA, V, VI, and part of VII were conducted with no need for Pringle maneuver (Fig. 1E). There was no need for transfusion intraoperatively. The patient recovered excellently from the operation and was discharged at the 8th postoperative day. Histological examination of the tumor revealed high grade sarcoma consisting mainly of spindle and focally of large pleomorphic cells with severe nuclear atypia, many mitoses and focal coagulative necrosis. Spindle cells revealed mainly fascicular arrangement. Immunohistochemically the tumor cells are positive for Vimentin, smooth muscle actin (SMA), focally for Caldesmon and are negative for Desmin, S100, CD34, CD117, WT1 and ER (Fig. 2). Since final diagnosis was leiomyosarcoma the patient was advised to receive adjuvant chemotherapy. The patient refused to receive adjuvant chemotherapy, and revealed evidence of recurrence six months after the operation, and finally died 12 months after the operation and 16 months after initial diagnosis.
DISCUSSION
Leiomyosarcomas are malignant neoplasms that arise from smooth muscle. Hepatic leiomyosarcoma may arise from intrahepatic vascular structures, bile ducts or ligamentum teres.456 Tumors arising from hepatic veins may develop Budd-Chiari syndrome and have worse prognosis with tumors arising from the ligamentum teres with better prognosis due to its increased resectability.4 Clinical manifestations of PHL are non-specific, patients are asymptomatic until tumors increase significantly in size. Most common symptoms are abdominal discomfort or pain, weight loss. In physical examination hepatomegaly and palpable mass are the most common findings. Liver function tests (LFTs) are usually within normal range and the same applies for AFP and CEA or CA 19-9. Histological preoperative diagnosis of primary hepatic leiomyosarcoma is controversial as with other liver tumors CT findings of PHL usually reveal a large, well-defined and heterogeneous hypodensity mass with internal and peripheral enhancement,789 and this was consistent with our CT findings. Histopathological diagnosis of PHL is characterized by presence of uniform and diffuse infiltrates of spindle shaped cells with hyperchromatic nuclei. To further confirm the diagnosis, a positive immunohistochemistry reaction for SMA, desmin and vimentin, as well as a negative reaction for keratin and S 100 protein is essential.1011 In our case, a microscopic examination revealed intersecting bundles of spindle shaped cells with eosinophilic cytoplasm and nuclear atypia. Additionally, immunohistochemistry revealed a positive reaction for SMA and desmin, and a negative reaction for c-kit receptor (CD117), DOG1, HMB45, PNL2 and Ki 67. Therefore, the diagnosis of PHL was confirmed.
Primary hepatic sarcomas and especially PHL are rare tumors, evidence is limited, and standard of care could not be safely defined. However, surgical resection followed by adjuvant chemotherapy is being widely accepted and practiced.2 Liver resection surgery forms the cornerstone of successful management of primary hepatic leiomyosarcoma with intention of R0 resection. It could be advised that all patients with potentially resectable tumors with adequate remnant liver volume should undergo surgical exploration and liver resection.
Extrahepatic tumor spread, diffuse intrahepatic tumor and impaired liver function are the main contraindications for surgery. Liver transplant has been reported in primary hepatic leiomyosarcoma, but the indication is weak compared to other liver tumors or even in the case of primary hepatic epithelioid hemangioendothelioma.12 The outcome for liver transplant in PHL varied with most of the cases developing recurrent or metastatic disease.12
The role of adjuvant chemotherapy/chemo radiotherapy is not well defined. The rarity of the disease precludes randomized trial and all recommendations are based on personal expertise. Most of the patients receiving R0 liver resection are referred for adjuvant chemotherapy. In our case the patient had R0 liver resection and was referred to the oncology department. She was advised to receive adjuvant chemotherapy but refused repeatedly.

 XML Download
XML Download