

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Although non-Hodgkin's lymphoma (NHL) commonly involves the liver in more advanced stages, primary involvement of the liver in this disease is rare.1 Primary hepatic lymphoma (PHL) is an extremely rare malignancy, representing <1% of all extranodal lymphomas.2 Clinical manifestations, laboratory findings, and radiologic features of PHL are non-specific. Therefore, PHL might be misdiagnosed as primary hepatic tumor or metastatic hepatic tumor.3 Although most patients are treated with chemotherapy, various approaches including surgery and radiotherapy have been attempted for better outcome. The optimal treatment of PHL remains unclear and its prognosis is poor.4 We report a case of PHL in a patient who had a mass-forming intrahepatic cholangiocarcinoma preoperatively. After surgical resection followed by chemotherapy, he has maintained long-term remission.
CASE
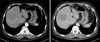
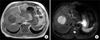
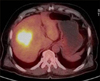
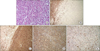
A 65-year-old man was transferred to our hospital for further evaluation of a liver mass that had been detected by ultrasound. The patient presented with abdominal discomfort history for one week. Past medical history was unremarkable. He had not experienced fever, night sweating, or other specific symptoms. On admission to the hospital, physical examination revealed no palpable mass, splenomegaly, hepatomegaly, or any lymphadenopathy. Initial laboratory findings showed normal complete blood counts and normal levels of bilirubin and transaminase. His serum alkaline phosphatase (ALP) level was in the upper range of normal (329 IU/L; normal range, 104-338 IU/L), so was his lactate dehydrogenase (LDH) level (240 IU/L; normal range, 130-270 IU/L). Serologic tests for hepatitis B virus (HBV), hepatitis C virus (HCV), or human immunodeficiency virus (HIV) were negative. Serum alpha-fetoprotein (AFP), carcinoembryonic antigen (CEA), and carbohydrate antigen 19-9 (CA19-9) levels were 2.0 ng/ml, 5.8 ng/ml, and 0.6 U/ml, respectively. A computed tomography (CT) scan revealed a lower density of the liver than that of the spleen, suggesting fatty liver. The scan also revealed a 6×6 cm sized slightly lobulated low-density mass in segment 8 with peripheral rim-like enhancement and mild dilatation of the peripheral intrahepatic duct. Splenomegaly or intraabdominal lymphadenopathy was not observed (Fig. 1). Magnetic resonance imaging (MRI) scan of the liver revealed low-signal intensity on T1-weighted image but high-signal intensity lobulating contoured mass on T2-weighted image (Fig. 2). With a suspicion of intrahepatic mass-forming cholangiocarcinoma, the patient underwent 18F-FDG positron emission tomography (18F-FDG PET) scan to check for metastasis. The scan showed a hypermetabolic lesion in segment 8 of the liver and focal hypermetabolic lesion in the colon (Fig. 3). To rule out the possibility of liver metastasis of gastrointestinal origin such as colon cancer, esophagogastroduodenoscopy and colonoscopy were performed. No specific findings were evident in the stomach or colon. It was diagnosed as intrahepatic mass-forming cholangiocarcinoma. Therefore, we decided to perform surgical resection. Indocyanine green retention test at 15 minutes (ICG-R15) was 14.8% and remnant liver volume in case of performing right hepatectomy was 24.8%, which prompted us to perform right anterior sectionectomy. Gross finding of the resected liver specimen is shown in Fig. 4. The tumor was a well-defined mass at 6.5×5.5 cm in diameter. Histologic examination revealed diffuse proliferation of lymphoid cells. Immunohistochemical staining revealed that these tumor cells were negative for hepatocyte marker, positive for leukocyte common antigen (LCA), positive for CD20, and negative for CD3 (Fig. 5). These histologic findings confirmed non-Hodgkin's diffuse large B-cell lymphoma. Bone marrow biopsy was done to confirm PHL. Normal bone marrow was detected. The tumor was confined to the liver without evidence of lymphomatous involvement in lymphoid structures, confirming PHL. The patient was given six cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone) chemotherapy from the 16th postoperative day, followed by maintenance cyclophosphamide therapy for one year. At follow-up of 80 months, the patient remains disease-free.
DISCUSSION
Diagnosis of PHL for this patient was based on several criteria. First, at the time of disease presentation, the patient's symptoms were mainly caused by liver involvement. Second, there was an absence of palpable lymphadenopathy. In addition, there was no radiological evidence of distant lymphadenopathy. Third, there was an absence of leukemic blood involvement in the peripheral blood smear. Patients with splenic, lymph node, or bone marrow involvement can be excluded from PHL diagnosis.45 Although the liver contains lymphoid tissue, host factors can make the liver a poor environment for the development of malignant lymphoma. Therefore, PHL is rare, accounting for <0.4% of extranodal NHLs and 0.016% of all NHLs.467 It can occur at any age. However, it is usually found in the fifth or sixth decade of life with a male-predominance.6
The pathogenesis of PHL remains unclear, although viral infections such as HBV, HCV, Epstein-Barr virus, human T-lymphotropic virus, liver cirrhosis, systemic lupus erythematous, and immunosuppressive therapy have been implicated.26 Hepatitis C is found in 40–60% of patients with PHL. Possible roles of HCV, cirrhosis, and therapeutic interferon in lymphomagenesis remain hypothetical.24 Conflicting theories exist on the association between HBV infection and PHL.24 Aozasa et al.8 have postulated that chronic antigenic stimulation by HBV might play a role in the development of PHL. Although it remains uncertain to what extent HBV contributes to the development of PHL, a host environment with impaired immunity might play an important role.24 However, our patient had none of the above conditions or risk factors for PHL.
Clinical manifestations of PHL are generally non-specific. The most common symptom is abdominal pain. Other symptoms can include weight loss, fever, anorexia, nausea, night sweats, and vomiting. Hepatomegaly is found in most (75–100%) patients. Symptoms such as fever, night sweats, and weight loss are present in 37–86% of patients, which jaundice is only present in 4% of patients.269 Hypercalcemia is found in 40% of patients.2 The reason for such results is currently unknown. Liver function tests, including transaminases, ALP, LDH, and bilirubin are usually elevated in PHL patients. LDH activity, a well-established lymphoid tumor marker, is increased in 87% of PHL patients.9 Tumor markers are usually within normal ranges, with AFP and CEA being normal in virtually all patients. Elevated LDH with normal AFP and CEA is a valuable biologic feature of PHL.6 However, in this study, our patient did not have abnormal levels of these parameters in laboratory findings or tumor markers.
Imaging presentation of PHL is variable, including solitary intrahepatic lesion, multiple nodules, and diffuse infiltration of the liver. However, sometimes there are only signs of homogeneous hepatomegaly.39 In an analysis of nine PHL patients, ultrasound performed in all patients demonstrated hypoechoic lesions to the surrounding normal liver parenchyma in 88.9% of cases.4 On CT, PHL lesions usually appeared as hypoattenuating lesions. They might have a central area with low intensity that indicates necrosis. Following the administration of intravenous contrast agent, PHL lesions might show slight enhancement. Although findings are variable, hypo-intensity on T1-weighted image and hyper-intensity on T2-weighted image have been described for PHL lesions.4
Due to the rarity of this disease entity and its non-specific clinical presentation and laboratory and radiologic features, a definite clinical diagnosis of PHL is difficult. PHL may be confused with hepatitis, primary hepatic tumors, hepatic metastases from gastrointestinal carcinoma, and systemic lymphoma with secondary hepatic involvement.2 Normal levels of tumor markers AFP and CEA are found in almost 100% of patients with PHL. Thus, the diagnosis of PHL requires liver biopsy compatible with lymphoma and the absence of lymphoproliferative disease outside the liver.2 Immunohistochemical studies with specific antibodies are essential to obtain correct diagnosis of PHL.9 In the patient described in this case report, tumor cells were stained positive for LCA but negative for hepatocyte marker, confirming the diagnosis of lymphoma. In addition, findings of CD20 positivity and CD3 negativity suggested that the tumor originated from a B cell. About 80% of PHL cases involve B cell tumors, with T cell phenotype accounting for only 8 to 28% of cases.10
Most patients are treated with chemotherapy. Some physicians employ a multimodality approach that incorporates surgery and radiotherapy. The optimal treatment of PHL remains unclear. The role of surgery is not fully clarified yet. However, liver resection followed by adjuvant chemotherapy and/or radiation has been reported to result in better prognosis. A small retrospective analysis of nine patients with PHL treated with liver resection has shown that surgery followed by chemotherapy has a better outcome.4 In addition, postoperative chemotherapy has better outcome.4 Moreover, postoperative chemotherapy is found to be the only prognostic factor for survival.4 In patients with localized disease, surgery followed by adjuvant chemotherapy should be considered to prevent disease recurrence.5 Similarly, a large review of the literature has shown that patients treated with surgery followed by chemotherapy have better survival rates.9 The standard treatment for patients with diffuse large B-cell lymphoma is the CHOP regimen. The addition of rituximab, a monoclonal antibody targeting pan-B-cell antigenic marker CD20, to the CHOP regimen, can augment complete response rate and prolong event-free survival and overall survival in elderly patients with diffuse large B-cell lymphoma without clinically significant increase in toxicity when it is given for 8 cycles. For patients with diffuse large B-cell NHL, several large-scale prospective randomized trials have demonstrated prolonged remission when rituximab is incorporated into the first-line treatment.231112 Whether systemic chemotherapy alone will give results comparable to surgery in resectable cases is currently unclear. The optimal treatment for PHL may continue to be elusive.9
In conclusion, PHL is a rare disease. It is easy to misdiagnose it due to the lack of specific clinical manifestations, imaging findings, and biochemical indicators. When space-occupying liver lesions with elevated LDH and normal AFP and CEA are found, PHL can be considered. PHL can easily be mistaken for other malignancies. To provide distinction, liver biopsy for immunohistochemical staining should be performed. If the diagnosis is made, early aggressive combination of chemotherapy should be started. The role of surgery has not been clarified yet. In select patients with localized tumor lesion, surgery followed by adjuvant chemotherapy should be considered for better outcome.
 XML Download
XML Download