

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Aneurysms are vascular pathologies that arise as abnormal expansion in a portion of an artery due to focal weakness within the vessel wall. The etiology of aneurysms is poorly understood, however, aneurysmal degeneration appears to be a multifactorial process resulting from changes in hemodynamic conditions and alterations in vascular wall biology that lead to loss of structural proteins and wall strength with subsequent dilation.
The two most common aneurysms are cerebral aneurysms (CAs) and abdominal aortic aneurysms (AAAs). Rupture of these aneurysms is a major source of morbidity and mortality. Ruptured CAs are the leading cause of non-traumatic subarachnoid hemorrhage,44)129) and ruptured AAAs are the 13th overall leading cause of death in the United States. The high burden of morbidity is the basis for current ongoing research to understand the underlying mechanisms of disease and developing technologies to prevent aneurysmal rupture.
Traditionally, efforts to further understand and treat CAs and AAAs have been conducted by separate groups of different disciplines. The two pathologies are rarely viewed in the same category. Despite anatomical differences, we believe that there are some similar and intersecting pathways for the pathological mechanisms at play. In addition, some differences between CAs and AAAs can yield further interesting insight into the unique hemodynamic effects that result in pathology. By conducting a thorough and focused review of the two topics, we aim to create a review that critically compares the two pathologies, while highlighting similarities that can broaden understanding of vascular pathology.
CLINICAL OVERVIEW
Based on estimates, 3.5-6.5% of the population over age 30 harbors an unruptured CA.9)85)94)123)125) CA rupture results in subarachnoid hemorrhage (SAH), which has a mortality of 40-50%,42)94) and more than half of survivors are left disabled.29)46) CAs can be classified according to three groups, based on size: small with diameters less than 10 mm, large with diameters of 10-25 mm, and giant with diameters larger than 25 mm. There are, however, many other ways to classify CAs.9)
AAAs are a relatively common vascular pathology with estimated incidence ranging from 5-9% in patients older than age 50.6)20)88) Patients harboring an AAA are asymptomatic until the aneurysm ruptures, resulting in severe morbidity and mortality.20) AAAs are defined as localized dilations of the abdominal aorta that exceed the normal diameter of the aorta by greater than 50%. AAAs expand at rates up to 0.25-0.75 cm per year, initially slower, then faster as they become larger.10) If not treated, many lesions will continue to enlarge until they rupture.44)
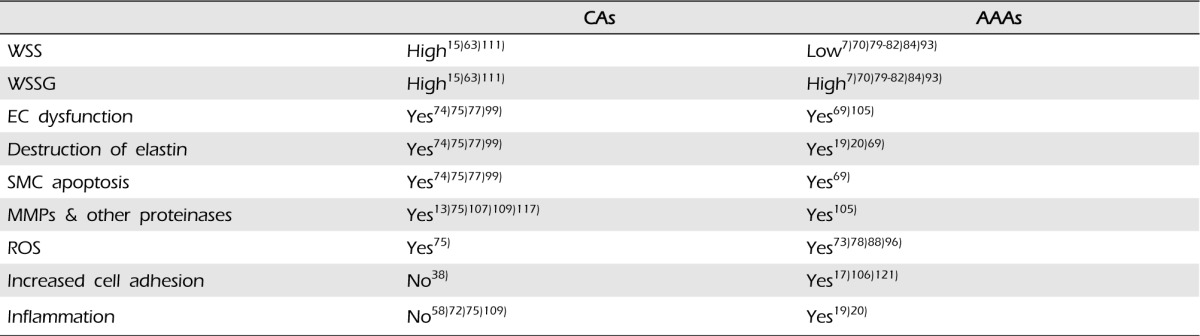
CAs and AAAs have many risk factors in common. Both are associated with older age, cigarettes, hypertension, and familial predisposition. However, these aneurysms have different gender prevalence. CAs are more common in females, with a nearly 2:1 female to male ratio,1)8)49)55)90)95)98) whereas AAAs are overwhelmingly more common in males, with a 4:1 male to female ratio.35)51)64)65) In addition, as described above, they have different morphologies and develop under different hemodynamic conditions. Most CAs arise as saccular (berry-like) dilation on the cerebral arteries of the circle of Willis under high blood flow, high wall shear stress (WSS), and high wall shear stress gradient (WSSG) conditions. AAAs arise as fusiform (spindle-like) dilations on the infrarenal aorta under low blood flow, low, oscillating WSS, and high WSSG conditions. Despite marked differences in their morphology and the structure of the arteries in which they form, CAs and AAAs share many histopathological and biomechanical characteristics.44)
HEMODYNAMICS
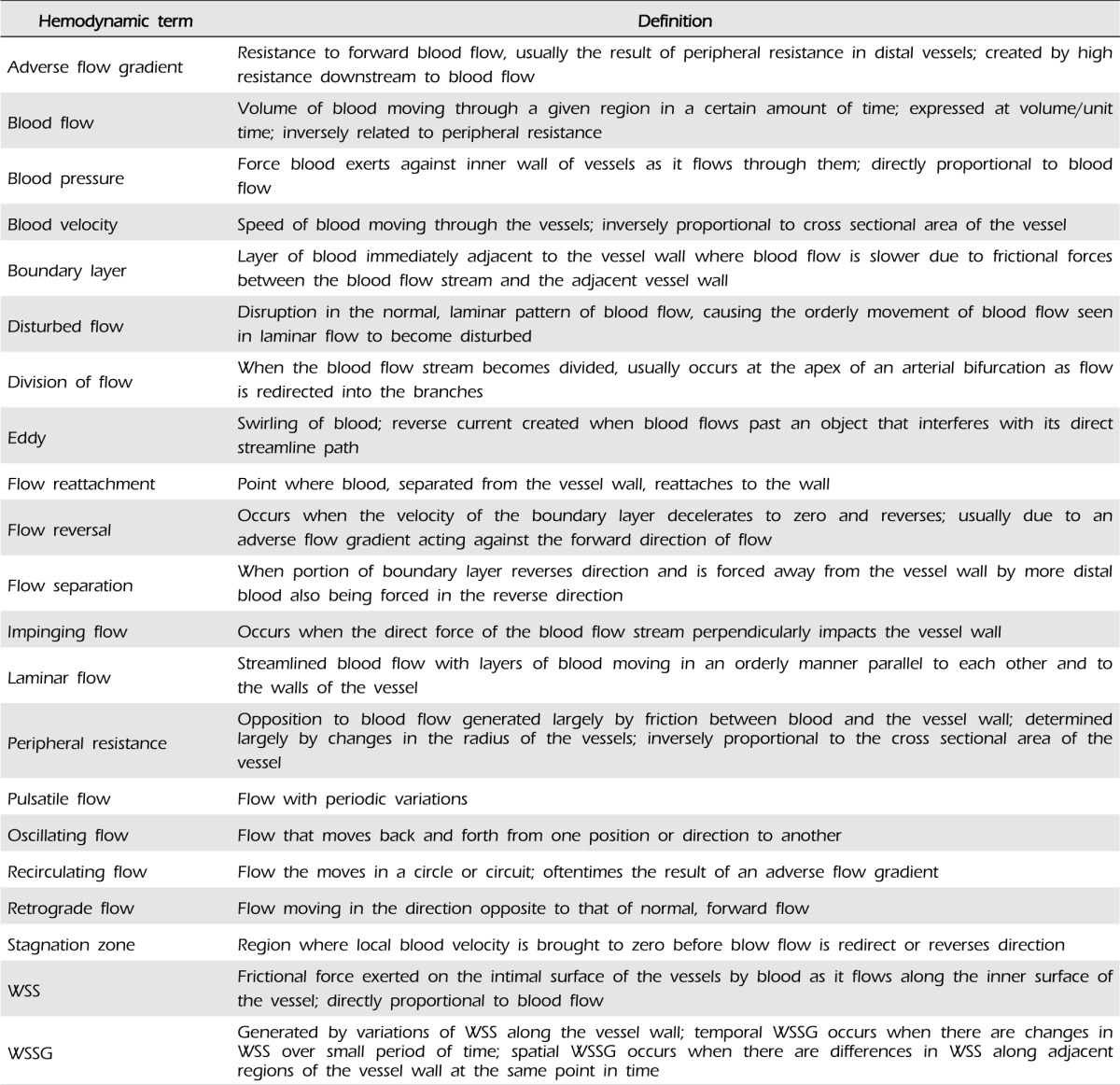
Hemodynamics is the study of blood flow and the factors that govern flow through the blood vessels. Heart contractions produce pulsatile changes in blood pressure and flow that fluctuate according to the cardiac cycle. Blood flow patterns and hemodynamic forces are not uniform throughout the vascular system. In straight segments of arteries, blood flow is anterograde and laminar.87) This is the normal pattern of blood flow throughout most of the circulatory system.87) However, blood flow is unsteady and may become disturbed at bifurcations, curvatures, or other regions with complex geometry, resulting in disorganized, irregular secondary flow patterns.31)87) Some of these patterns will be described as relevant throughout this paper. See Table 1 for terms related to hemodynamics.
Blood flow generates hemodynamic forces that act on blood vessel walls.86) Wall shear stress (WSS) is the frictional force exerted by blood as it flows along the intimal surface of the arterial wall.74)87)125) WSS is related to blood flow, such that increased flow leads to increased WSS and decreased flow leads to decreased WSS.
Anterograde, laminar flow provides a continuous, directed WSS, which is distributed uniformly along the vessel wall. When blood flow is disturbed, WSS is also disrupted, causing variation in the WSS along the adjacent wall. Temporal differences in WSS occur with increases or decreases in WSS over a short period of time at the same location and create an oscillatory shear index (OSI). Spatial differences in WSS between two close points of a cell layer at the same point in time create a wall shear stress gradient (WSSG), such as when flow is accelerating (positive WSSG) or decelerating (negative WSSG).75)
Accelerating flow (positive WSSG) creates a stretching force within the surface of endothelial cells (ECs) as frictional drag increases in the direction of flow.24)67)122) In contrast, decelerating flow (negative WSSG) can cause compression within the surface of ECs.24) These forces may augment or diminish the stretch already exerted across the ECs by the WSS generated by the blood flow.24)
Hemodynamics: cerebral arteries
The circle of Willis is the cross-circulation of cerebral arteries that supplies blood to the brain and surrounding structures.87) It is formed by the confluence of the right and left internal carotid arteries anteriorly and the basilar artery posteriorly. There is considerable normal anatomic variation within the circle of Willis.87) CAs predominantly arise along or within the vicinity of the circle of Willis, with over 85% of CAs occurring in the anterior part of the circle of Willis.9)63)
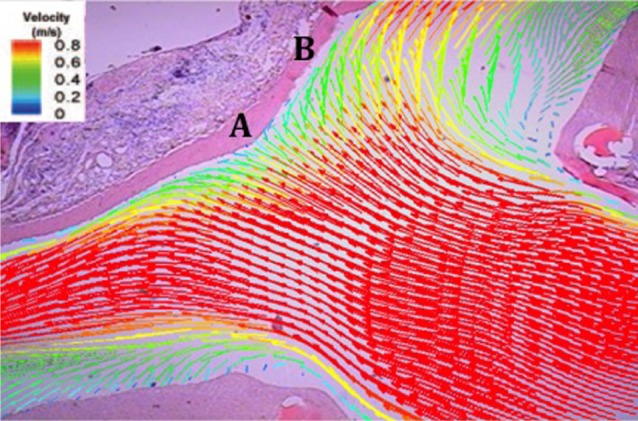
The cerebral arteries have small radii, thus, they are exposed to relatively higher baseline blood flow and WSS than other vessels.94) In addition, the tortuosity and sharp curvature of the circle of Willis give rise to a complex geometry and unique hemodynamic environment as blood flow can become disturbed in these regions.38)94)125) As mentioned above, CAs appear most frequently at the apex of bifurcations, but they are also found at the outer lateral wall of curved arterial segments.2)9)27)63)87)97)108)110) These areas are characterized by impinging blood flow with high WSS and high WSSG.63) Impinging flow occurs when the direct force of the blood flow stream acts perpendicular to the vessel wall. This results in a local pressure elevation where the blood flow stream impacts the vessel wall at the site of the bifurcation,9) as well as a high and spatially varying WSS downstream of the impingement point.75)
Bifurcations and the outer lateral wall of sharp curvatures act as flow dividers. When a vessel bifurcates, blood flow impinges at the apex of the bifurcation and divides as it is redirected toward the outer wall of the branches. Blood flow is fastest at the bifurcation and slowest near the outer wall of the proximal branch. Similarly, at sharp curvatures, the outer lateral wall acts as a one-way flow divider redirecting blood from the outer wall of the curvature toward the inner wall. At sharp curvatures, blood flow is fastest near this outer lateral wall where flow is divided and slowest near the inner wall of the curvature. In these regions, blood flow oftentimes becomes disturbed, giving rise to secondary patterns of flow.59)60)87)102)128) (Fig. 1)
As mentioned previously, CAs predominantly arise at bifurcations along the circle of Willis. The hemodynamic stresses at these regions are the result of impinging flow as well as high WSS and high WSSG.15)111) Impinging flow occurs when the direct force of the blood stream perpendicularly impacts the arterial wall. This impacting force results in a local pressure elevation on the arterial wall in the region of impact.9)27) As the bloodstream approaches the bifurcation, it impinges at the apex, and the apical wall divides the flow stream by deflecting blood towards the branches.63) This creates a central stagnation zone at the apex, where the local velocity is brought to zero at the apical wall as it is redirected,94) which creates a region of higher pressure immediately proximal to the apex as flow accumulates behind the stagnation zone, and a region of lower pressure at the entrances to the branches immediately distal to the apex. As a result of this pressure gradient, blood flow accelerates as it enters the branches to a maximum velocity, and then begins to decelerate toward baseline, physiologic velocity. In this acceleration zone, the already high WSS generated by the impinging flow at the apex becomes increasingly higher, with the highest WSS at the point of the maximum velocity.75) As a result of the accelerating flow, the arterial wall in the acceleration zone experiences a progressively increasing WSS along the direction of flow, and this large spatial gradient in WSS causes high, positive WSSG.92) This progressively increasing WSS gives rise to a large WSSG in the region immediately distal to the apex.75)
At high flow velocities, blood at the boundary of the arterial wall separates from this wall as it accelerates in the branch and reattaches downstream. If flow increases even more, a helical pattern of flow may develop in this pocket where flow separated from the wall. This further disturbs flow in this region and generates even larger variations in WSS, leading to a higher WSSG.92) These secondary patterns of blood flow generated by the bifurcation and sharp curvatures of vessels govern the local distribution of WSS and WSSG along the vessel wall in these regions.11)104)
Evidence for the role of disturbed hemodynamics in the pathogenesis of CAs
There is significant anatomic variation within the normal human circle of Willis.87) According to one report, up to 60% of people do not have a completely closed circle.63) The WSS experienced at a bifurcation is dependent on its geometry, including the radii of all vessels involved and the bifurcation angle.94) Thus, anatomic variations in vessel diameter, bifurcation angle, and local asymmetries have considerable effects on blood flow and WSS.87)125) This has the potential to create a hemodynamic environment that predisposes to aneurysm formation.52)63)94) Thus, the presence of anatomical variations, particularly those resulting in asymmetries along the circle of Willis, is thought to have a significant role in aneurysm formation by influencing the local hemodynamic environment.27)41)52)87)
Increased arterial flow to the cerebral arteries has been shown to exacerbate the already disturbed hemodynamic environment, and evidence suggests that this further contributes to the predisposition for CA formation in these regions. CAs are associated with vascular anomalies, such as fenestrations, persistent carotid-basilar anastomoses, internal carotid agenesis, and high-flow arteriovenous malformations, which result in increased blood flow to the cerebral arteries and/or asymmetries in the distribution of hemodynamic forces along the circle of Willis.9)14)66)91)103) CAs are commonly observed on arterial pedicles that feed arteriovenous malformations, which suggests that the increased arterial blood flow due to arteriovenous shunting provides a conducive environment for aneurysm formation.12) In addition, experimental studies using animals have demonstrated that CAs can be formed by altering the hemodynamic stresses along the circle of Willis.30)37)40)
Disturbed patterns of flow generated at the stagnation zone increase as the bifurcation angle becomes blunter, and studies have shown that CAs occur at higher frequencies at blunter bifurcation angles.4)125) Asymmetries in the daughter vessels branching from the bifurcation also appear to predispose to CAs.
Hemodynamics: Aorta
The aorta, the largest artery in the human body, is highly distensible with very low resistance to flow. As the heart ejects blood during systole, the aorta stretches to accommodate this large volume of blood, and while the heart is refilling during diastole, the aorta recoils, driving blood forward throughout the rest of the vascular system.
Aneurysms may develop at any location along the aorta, but the AAA is the most common aortic aneurysm. More than 90% of AAAs arise in the infrarenal segment of the abdominal aorta, located below the renal arteries and above the aortic bifurcation into the iliac arteries. The normal infrarenal aorta is approximately 12 cm long, 2 cm in diameter, and 2 cm thick.
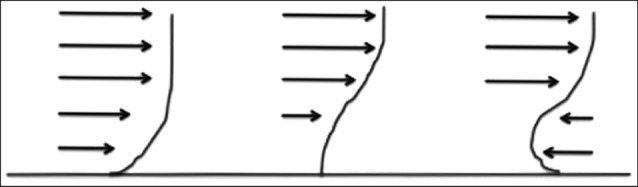
There is marked variation in the hemodynamic conditions along the length of the aorta, and this is thought to contribute to the predilection of aneurysms to form in the distal aorta.25) One of the most significant differences is between the resting aortic WSS in the suprarenal and infrarenal segments of the aorta.25) In the suprarenal aorta, flow is anterograde and laminar throughout the entire cardiac cycle, providing continuous, directed WSS. In the infrarenal aorta, flow is anterograde at the beginning of the cardiac cycle, however, toward the end of systole and throughout diastole, there is reverse, recirculating flow and low, oscillating WSS.25) (Fig. 2)
The disturbed flow patterns observed in the infrarenal aorta during the latter part of the cardiac cycle are the result of high peripheral resistance in the arteries that supply blood to the lower extremities.116) During resting conditions, the muscles of the leg require little blood flow, so that there is high peripheral resistance to flow. These resistive hemodynamic conditions impose an adverse pressure gradient on the infrarenal aorta.116) An adverse pressure gradient occurs when the pressure increases in the direction of flow, and it essentially opposes the forward flow.
At the beginning of systole, when blood pressure is the highest, flow is able to overcome the adverse pressure gradient and continue moving forward. As blood pressure decreases at the end of systole and throughout diastole, the adverse pressure gradient begins to retard flow. The effect of this adverse pressure gradient is felt most strongly near the arterial wall in the layer of flow at the boundary of the vessel wall where flow is already slower due to friction from the adjacent arterial wall. At some point, the adverse pressure gradient slows the flow velocity at the boundary layer to zero and then reverses the flow. This causes flow separation as the boundary layer of the immediately upstream forward flow is diverted away from the wall as it flows around the reversed flow.26)100) As separated flow moves past the reversed flow, it reattaches to the arterial wall downstream. The flow separation and reattachment divides flow into a recirculating flow and a downstream flow with its boundary layer passing over the region of recirculation. The region of recirculating flow is sometimes referred to as the recirculation bubble. With the start of the next cardiac cycle, blood pressure increases and is able to overcome the adverse pressure gradient, and the recirculating flow reverses directions and flows in the forward direction again.
The adverse pressure gradient leads to deceleration of blood in the infrarenal aorta and, subsequently, results in a low WSS. The flow reversal as well as separation and reattachment of flow create large differences in WSS along the infrarenal aortic wall. In addition, the pulsatile nature of the blood flow generates oscillations in flow with WSS moving back and forth in direction during different parts of the cardiac cycle. These flow patterns give rise to high spatial and temporal WSSG experienced by the vessel wall in the infrarenal aorta.
Thus, development of AAAs occurs under conditions of high peripheral resistance, which leads to diminished anterograde flow in the distal aorta. This creates a unique hemodynamic environment characterized by disturbed, recirculating flow and low, oscillatory WSS and high WSSG.7)70)79)80)81)82)84)93)
Evidence for the role of disturbed hemodynamics in the pathogenesis of AAAs
As described above, AAAs have a strong predilection to form under the disturbed, oscillatory flow and low WSS environment of the infrarenal aorta. AAAs are associated with various conditions, such as major limb amputations, spinal cord injury (SCI), and peripheral artery occlusive disease, that alter and/or reduce blood flow to the lower extremities.32)101)118)119)127) Narrowing of peripheral arteries, due to reduced blood flow demand (long resting periods in patients with SCI) or total occlusion of arteries, increases the resistive hemodynamics of the peripheral arteries, which increases the adverse pressure gradient in the aorta exacerbating the disturbed, oscillatory flow and low WSS environment in the infrarenal aorta.116)130)
Patients with major lower limb amputations are five times more likely to develop AAAs compared with non-amputee patients, likely secondary to ligation of the distal femoral artery.120) Interestingly, the morphology of the aneurysms in amputee patients appears to be strongly influenced by the laterality of the amputation. This supports the observation that chronically decreased and/or altered blood flow in the distal aorta contributes to the pathogenesis of AAAs.
Patients with SCI have been studied as an extreme example of chronically decreased blood flow to the lower extremities. These studies have demonstrated an association of SCI with increased distal aortic diameter and narrowing of the iliac arteries. These patients have significantly diminished anterograde flow and WSS in the distal aorta throughout the cardiac cycle,25) which suggests that these disturbed hemodynamic conditions contribute to the pathogenesis of AAAs. In addition, the narrowing of the iliac arteries supports that chronically decreased blood flow results in inward vascular remodeling and narrowing of the iliac arteries and other arteries of the lower extremities.113)127)
Exercise is associated with a decreased risk of AAAs.25) Exercise increases blood flow to the lower extremities, reducing the resistive hemodynamics of the peripheral arteries.116) It has been shown that as blood flow increased during exercise, the retrograde, oscillatory flow and WSS observed in the infrarenal aorta during rest disappeared.115) Studies have shown that, even with a modest amount of activity, blood flow in the distal aorta remains anterograde throughout most of the cardiac cycle, nearly eliminating retrograde and oscillatory flow. This suggests that exercise may exert its protective effects against AAAs by diminishing hemodynamic conditions associated with aortic pathology.25)
WSSG as a common thread
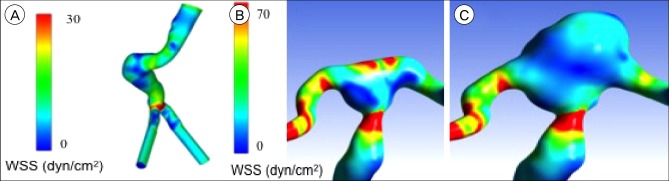
Clearly, CAs and AAAs exist under very unique hemodynamic circumstances as well as anatomically and geometrically different locations. Although the two pathologies exist at the opposite ends of the WSS spectrum (See Fig. 3), they both experience high WSSG. These changes in blood flow have been shown to be a critical factor in inducing arterial remodeling.38)42) Through a process called vascular remodeling, arteries adapt and change in response to variation in their hemodynamic environment.38) Vascular remodeling usually occurs as an adaptive process by which the vessel undergoes alterations in size and/or composition in order to maintain vascular homeostasis; however, in some circumstances, unusual and maybe chronic hemodynamic conditions may cause an abnormal, or pathological, biological response. This abnormal response is hypothesized to play an important role in the pathogenesis of aneurysm.38)87) The process of vascular remodeling is thought to be a fundamental part of many vascular diseases.31)
MECHANOBIOLOGY AND THE CELLULAR RESPONSES OF DISTURBED HEMODYNAMICS
Aneurysm formation involves processes that cause degradation of the structural components and loss of mechanical integrity of the arterial wall.89) This is largely targeted at degradation of elastin as this permits the initial dilation of the vessel.19) Loss of elastin is thought to represent an early step in the pathogenesis of aneurysms, as this permits the initial bulge in the wall.58) Subsequently, there is loss of smooth muscle cell (SMC)s and a dynamic turnover in collagen.44)
The exact mechanism of aneurysm initiation remains unclear, but evidence suggests that an initial insult to the arterial wall may trigger a pathological chain of events that ultimately results in the development of an aneurysm. Once this initial insult occurs, the process of aneurysm development appears to be self-sustaining.74) A growing body of evidence supports the role of disturbed hemodynamics as a critical component in the initiation of aneurysms.16)74) EC damage is a universal pathological finding in both CAs and AAAs, and although very different from each other, it is possible that the unique hemodynamic environments in which CAs and AAAs arise cause mechanical damage to the arterial wall through EC loss or dysfunction. This mechanical damage alone may not be enough to initiate aneurysm formation, but it may ignite a cascade of downstream events leading to development of aneurysms.
Blood flow and the hemodynamic forces that it exerts on the surrounding vessel wall have a pivotal effect on the vasculature.21)71)99)112) ECs are constantly exposed to blood flow and the hemodynamic forces it generates. Through a process called mechanotransduction, ECs sense their hemodynamic environments by converting mechanical stimuli into biochemical signals that modulate the structure, composition, and function of the various components of the vessel wall.43) ECs respond to alterations in these hemodynamic forces, specifically WSS, by initiating a process of vascular remodeling in order to return WSS to its baseline value.23)31)47)48)74)
Proposed mechanisms of pathogenesis for CAs
Most CAs arise as saccular dilations composed of a thin-walled sac connected to the parent vessel by an orifice called the neck.9) The aneurysm sac typically points in the direction blood would flow in the absence of a bifurcation.2)58) They are characterized by disruption of the ECs, degradation of the internal elastic lamina, and SMC loss with thinning of the tunica media,3)28)39)50)54)57)74)75)83)109)124)126) with locally increased metallomatrix proteinases (MMPs) being responsible for the degradation.53)72)122) The wall of CAs appears to progressively transform from nearly intact tissue at the neck to severely degenerated at the fundus.58)117) Wall thinning and SMC loss is the most prominent at the fundus, which may be completely lacking SMCs. In addition, there is decreased collagen and fibronectin within the wall, and collagen that is present appears disorganized.9)16)27) Increased MMPs have been found in the walls of CAs.13) Oftentimes, thrombus is observed within the aneurysm sac.9)
Experimental studies with animals as well as human autopsy studies have observed that the initial nidus of the CA is, oftentimes, located slightly distal to the fundus.9)117) As the aneurysm sac grows, the bifurcation becomes involved as well,117) which suggests that CAs initiation occurs at the most distal part of the acceleration zone where WSS and WSSG are the greatest.75)99)109) Studies have shown that the presence of high WSSG in addition to high WSS may have damaging effects on the vessel wall that lead to destructive, pathological remodeling.17)56)61)62)75)99) Specifically, this combination of hemodynamic factors has been shown to cause mechanical damage to ECs, leading to EC loss and dysfunction.74)77)99) EC damage may impede the ability of the arterial wall to respond normally to changes in its hemodynamic environment.16)75) The impact of this hemodynamic environment on the vessel wall may represent a fundamental step in initiating the cascade of events that leads to CA development.61)75)77)
High WSS induces nitric oxide (NO) secretion by upregulating endothelial nitric oxide synthase (eNOS).76)114) Under high WSS conditions, eNOS stimulates outward vascular remodeling.23)114) Interestingly, however, studies have shown that under conditions of high WSS and high WSSG, when ECs are damaged, as they appear in CAs, this results in loss of eNOS expression, impairing production and secretion of NO by ECs.46) It is possible that EC damage is a result of either chronic exposure to persistent, high WSS or to the coexistence of high WSS and WSSG. This reduces NO bioavailability, and under high WSS conditions, the reduction in NO stimulates SMC expression of iNOS to produce NO. In addition, after de-endothelialization, most luminal smooth muscle cells of the neointima are in contact with blood flow and express inducible nitric oxide synthase (iNOS) in vivo, and in vitro studies have shown that this is in response to exposure to WSS.33)122) The iNOS isoform, however, produces much larger amounts of NO compared with the eNOS isoform. These excessive amounts of NO are thought to react with the reactive oxygen species (ROS), causing oxidative damage to ECs and other components of the arterial wall.
Inflammatory cells are observed in the wall of CAs, but there is no evidence to suggest that inflammatory cells are present during the initiation of aneurysm.53)58)72)75)109) Although alterations in WSS, both high and low, are associated with increased expression of surface adhesion markers and EC permeability, adhesion of inflammatory cells requires both the presence of adhesion molecules on the surface as well as the residence time that the circulating inflammatory cells spend in any one region of the arterial wall.38) Thus, the high flow environment experienced by the cerebral vessels prevents inflammatory cell adhesion and infiltration by decreasing the residence time. Additionally, because NO is a potent anti-inflammatory agent that inhibits adhesion of circulating inflammatory cells to the EC surface, the excessive NO probably contributes to this absence of inflammatory cells at initiation of aneurysm.
Interestingly, a recent study using a rabbit model found that, despite absence of macrophages and other inflammatory cells during CA initiation, MMPs were increased. This study found that MMPs co-localize with SMCs, suggesting that SMCs are responsible for MMP production during CA initiation. Thus, under high WSS, and high WSSG during CA initiation, SMCs appear to behave as pro-inflammatory cells by secretion of MMPs necessary for CA remodeling.72)
Progressive SMC loss and the resulting medial thinning is another prominent feature of CAs.74)75) SMC loss is believed to be a combination of decreased SMC proliferation and increased SMC apoptosis. Normally, under high WSS conditions, transition of SMCs from a contractile phenotype to a more proliferative one occurs, resulting in production of ECM proteins, growth factors, and proteases that are important for remodeling of the vessel wall. Indeed, under high WSS and high WSSG, we have observed a loss of SMC contractile phenotype,72) but these altered SMCs do not appear to be synthetic, as cell proliferation is actually decreased and apoptosis increased,53)75) resulting in SMC loss. However, they do exhibit pro-inflammatory behavior, upregulating NF-κB and MCP-1 as well as producing MMP-2 and -9, which has been hypothesized as a result of EC signaling under such hemodynamic conditions.72) In addition, proliferation of SMCs is inhibited by NO, and it is likely that excessive NO contributes to SMC loss by inhibiting proliferation and inducing apoptosis.68)94) In addition to decreased proliferation, production of ECM proteins and other important factors by these SMCs is reduced, which further contributes to degeneration of the arterial wall as degraded ECM components are not replaced.
Proposed mechanisms of pathogenesis for AAAs
AAAs arise as fusiform dilations with an enlarged and elongated course, which circumferentially involves the parent vessel with no defined neck.9) They involve extensive structural remodeling of the entire circumference of a segment of the aorta, and are characterized by degradation of elastin and other ECM proteins, loss of SMCs, and abnormal collagen.69) Other pathologic features of AAAs include intense inflammatory infiltrate, consisting mostly of macrophages and leukocytes, and increased expression of MMPs.69) In greater than 90% of AAAs, an endoluminal thrombus is found attached to the wall of the aneurysm sac.36) In addition, atherosclerotic lesions are frequently found in association with AAAs, however, unlike in the past, the role of atherosclerosis in the pathogenesis of AAAs is highly controversial.
The intense infiltration of inflammatory cells, predominantly macrophages and lymphocytes, into the arterial wall is a striking feature of AAAs.20) This inflammation may be a response to changes in the vessel wall in response to low, oscillatory flow and WSS environment in the infrarenal aorta.20) Migration of leukocytes into the arterial wall is thought to stimulate secretion of chemokines, which may further aneurysm formation.20) The content of both elastin and collagen is decreased in AAAs, but the collagen to elastin ratio is increased, suggesting that as elastin is degraded, it is initially replaced by collagen. This reduces the elasticity of the aortic wall, leading to stiffening and decreased wall motion.18)34)
There is a growing body of evidence to support the role of disturbed hemodynamics as a critical component in the initiation of AAAs. The low, oscillatory WSS and high WSSG have been shown to cause damage to ECs, resulting in disturbance and/or loss of their regulatory functions. This may represent a fundamental step in initiating the cascade of events that leads to dilation.20,21,22)
Low, oscillatory flow and WSS predispose the arterial wall to inflammation by stimulating inflammatory cell adhesion and migration into the arterial wall.106) Low WSS increases EC permeability to the circulating blood elements and causes upregulated EC expression of pro-inflammatory molecules and surface adhesion factors. Independent of WSS, low flow increases the residence time that circulating blood elements spend near any region of the EC surface, which greatly facilitates adhesion to the ECs.17)121)
Once inflammatory cells enter the wall, they release ROS and MMPs, which degrade elastin and other components of the ECM. It has been suggested that elastin degradation products function as chemokines, recruiting more inflammatory cells to the wall. Inflammatory cells also release a cascade of cytokines that amplify the inflammatory response by activating chemokines and other pro-inflammatory factors, which recruit more inflammatory cells to the wall. Cytokines also promote increased synthesis and activation of MMPs and higher production of ROS. Migration of inflammatory cells may actually lead to degenerative changes in the wall that directly result in dilation of the vessel; however, the stimulus for these inflammatory cells may result from the abnormal hemodynamic forces that cause damage to ECs.
Common pathways for AAAs and CAs.
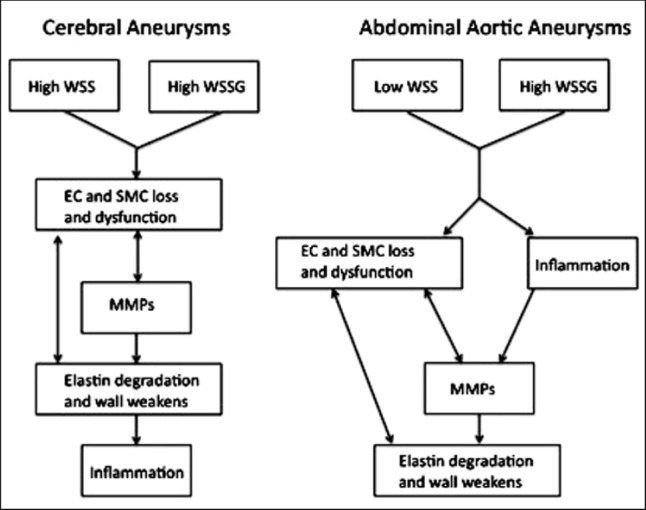
With WSSG as a possible inciting factor, a variety of mirroring pathways take place in the pathogenesis of AAAs and CAs. As mentioned above, EC's play a pivotal role in detecting the disturbed hemodynamics and help initiate the cellular events. Interestingly, although inflammation plays a very strong role in early stages of AAA pathogenesis, it seems to be a later consequence in the CA pathway. See Fig. 4, for a schematic of pathogenesis for AAAs and CAs. In addition, Table 2 summarizes events that are either common or unique to the vascular pathologies.
CAs and AAAs have traditionally been studied and treated by different groups of specialists. Common pathways they share are frequently overlooked or understated. This is evident in many aspects of the disease process, including treatment. While endoluminal reconstruction through an endovascular route has been the mainstay treatment of AAAs for decades, this has only recently been attempted in CAs.5)45) As further developments are made in both fields, particularly for cellular and molecular guided therapies, cross-disciplinary reviews will help broaden the understanding of the field.
 XML Download
XML Download