

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Gorlin and Goltz in 1960 defined the condition as a syndrome comprising the principal triad of multiple basal cell nevi, jaw keratocysts, and skeletal anomalies.1-3 The condition is also known as Gorlin syndrome, nevoid basal cell carcinoma syndrome, and basal cell nevus syndrome.4-7 Gorlin-Goltz syndrome is an autosomal dominant inherited condition that exhibits high penetrance and variable expressivity, however this disorder can arise spontaneously.2,7-9 Almost 60% of the patients with Gorlin-Goltz syndrome have no known affected family members.2 The Gorlin-Goltz syndrome gene has been mapped to chromosome 9q22.3-q31.5,7 The prevalence of Gorlin-Goltz syndrome has been estimated about 1 per 60,000.10 Males and females are equally affected.11 The main clinical features of Gorlin-Goltz syndrome includes multiple keratocystic odontogenic tumor (75%), basal cell nevi (80% in whites and 38% in blacks), and skeletal anomalies (70%).2,12 We present a report of Gorlin-Goltz syndrome with familial pattern affecting father and daughter.
Case Report
A 39-year-old male patient reported to the dental institute with the complaint of pus discharge from right lower posterior region of the jaw since one month ago. The patient noticed decayed teeth in the same region, which was not associated with pain. He had visited a private dental clinic, where decayed tooth had been extracted and medications had been given for about 5 days.
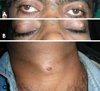
On general physical examination, the patient was well built, 182 cm tall with normal gait and satisfactory vital signs. The extraoral examination revealed hypertelorism, strabismus, and a cystic swelling on the left eyelid as well as his neck (Fig. 1). Intraoral examination revealed missing teeth of the right mandibular first and second molars and pus discharge from the same region. No other skeletal abnormalities were detected.
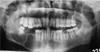
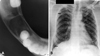
Panoramic radiograph revealed well defined multiple unilocular radiolucencies with sclerotic borders in the mandibular body, ramus, and symphysis areas. The unilocular radiolucencies varied in diameter from minimum 3 cm to around 7 cm in diameter. The largest one was located in the body of the mandible on the right side extending superiorly from the edentulous area to the lower border of the mandible inferiorly. Three smaller radiolucencies measuring around 3 cm in size were located in the posterior part of the right mandibular body and ramus as well in the left symphysis region. A smaller radiolucency measuring around 1 cm in size was observed in the periapical region of the right maxillary third molar. There was no radiographic evidence of tooth displacement and root resorption (Fig. 2). Cross sectional mandibular occlusal radiograph revealed a radiolucent area with minimum cortical plate expansion (Fig. 3A). The findings of panoramic radiograph raised the possibility of Gorlin-Goltz syndrome and further investigations were carried out. Chest radiograph revealed the bifid fourth and eighth rib on the right side (Fig. 3B).
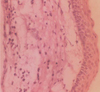
CT images showed hypodense areas in relation to the right mandibular body separated by a hyperdense septae. Minor breach in the cortical integrity was observed in the lingual aspect of the ramus region. A single lytic lesion was observed in relation to symphysis region. The lesion involved the right half of maxilla including maxillary sinus. Mucosal thickening in the right maxillary sinus was also observed (Figs. 4A and B). Calcification of falx cerebri and tentorium cerebella was also observed (Fig. 4C). Incisional biopsy of the lesion through intraoral approach revealed the following histopathological features: parakeratinized, corrugated, 6-10 layers thick epithelium with palisaded, polarized basal cell layer. The connective tissue showed daughter cysts suggestive of keratocystic odontogenic tumor (Figs. 5A and B).
Owing to the familial tendency of this diagnosed condition, the family members were subjected to thorough clinical and radiological examination. The patient's 8-year-old daughter showed a clinical feature of hypertelorism (Fig. 6A). Her panoramic radiograph revealed well defined, unilocular radiolucency measuring approximately 2.5 cm in diameter with sclerotic border in her mandibular symphysis region. The radiolucent area extended superiorly from the root apices of the mandibular anterior teeth to the lower border of the mandible inferiorly enclosing the left canine and premolar tooth buds (Fig. 6B).
Histopathological evaluation of the specimen obtained after intraoral biopsy procedure revealed histopathological features similar to that of her father (Fig. 7). No other family members were affected by this condition (Fig. 8). A diagnosis of Gorlin-Goltz Syndrome was made on the basis of clinical, imaging, and histological findings. The father and daughter were advised surgical removal of the keratocystic odontogenic tumor. Unfortunately due to certain logistical and financial reasons, the patients could not keep up with appointments.
Discussion
Gorlin-Goltz syndrome was first reported by Jarish in 1894.13 The syndrome has been designated by a variety of different terms including Gorlin syndrome, nevoid basal cell carcinoma syndrome, basal cell nevus syndrome, syndrome of jaw cysts, and jaw cyst - basal cell nevus - bifid rib syndrome.4-7,12 Gorlin suggested the term nevoid basal cell carcinoma syndrome, however all affected adults did not have basal cell carcinomas. The condition is known as Gorlin syndrome because of Gorlin's contributions to the understanding of the condition.7,12 Clinical features of Gorlin-Goltz syndrome arises at the first, second or third decade.2 In the present case, the features were identified at fourth decade and in daughter at the first decade. Gorlin-Goltz syndrome has an autosomal dominant mode of inheritance, but can arise spontaneously, or can have a variable phenotypic penetration.14 Similar pattern of inheritance was observed in our cases. Gorlin and Goltz in 1960 gave a complete description of the syndrome.1 This syndrome is associated with a wide spectrum of developmental anomalies and neoplasms.9 The most common and significant features are summarized in Table 1.
Keratocystic odontogenic tumor can be the first feature of the syndrome. The tumor is typically found as incidental radiographic findings. The tumor may manifest clinically if it become infected or cause symptoms such as swelling. The tumors were responsible for detection of the syndrome in our cases. Keratocystic odontogenic tumors in Gorlin-Goltz syndrome usually comprise unilocular or multilocular radiolucencies of the mandibular body, angle, or ramus.2 In children and adolescents, the cysts may cause displacement of developing teeth and delayed dental development.9 The present case also showed the similar radiological features such as the displacement of the tooth bud in the daughter's mandibular symphysis region. The most frequent skin lesions of Gorlin-Goltz syndrome are cutaneous basal cell carcinomas, benign dermal cysts, and palmar and plantar keratosis or pits.2,15 Apart from the cutaneous dermal cyst, there was no skin lesion in the father. Almost 70% of patients with this syndrome have some degree of cranio-facial anomalies. These can comprise frontal and parietal bossing and broad nasal root which may be associated with occular hypertelorism.2 Hypertelorism was also observed in these cases. Thoracic cage anomalies such as bifid and fused ribs may be present. Syndactyly or polydactyly of toes may occur.2 Although no digital abnormalities were observed in both of our cases, bifid ribs were observed in the father. Ectopic calcification of falx cerebri, tentorium cerebella, and bridged sella may also be detected radiologically.2 The diagnosis of Gorlin-Goltz syndrome requires the presence of two major or one major and two minor criteria.2,3,7,16 In the present cases, the following major and minor criteria were present: histologically proven keratocystic odontogenic tumors of the jaws, calcification of the falx cerebri, bifid ribs, and hypertelorism. The patients affected by Gorlin-Goltz syndrome must be evaluated by several relevant specialists to precisely confirm the diagnosis, detect the likely genetic basis, provide appropriate genetic counseling, and manage the various clinical manifestations. Early diagnosis and treatment may reduce the severity of the long term sequelae of Gorlin-Goltz syndrome including malignancy and oromaxillofacial deformation and destruction.




 XML Download
XML Download