

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Each year, the United States (US) Food and Drug Administration (FDA) receives several hundred thousand medical device reports (MDRs) of suspected device-associated deaths, serious injuries, and malfunctions [1]. Entries are submitted on the FDA's website and collected data are housed in the Manufacturer And User Facility Device Experience (MAUDE) database. Importantly, the MAUDE database aims to capture reports from all mandatory reporters, including manufacturers, importers, and users (e.g., hospitals, outpatient diagnostic and treatment facilities, nursing homes, ambulatory surgical centers). By the FDA's own admission, “… this passive surveillance system has limitations, including the potential submission of incomplete, inaccurate, untimely, unverified, or biased data,” though goes on to say that MDRs comprise only one of several important postmarket surveillance data sources [1].
In recent years, the placement of transvaginal surgical mesh (TVM) for the treatment of pelvic organ prolapse (POP) and stress urinary incontinence (SUI) in women has come under intense scrutiny by the FDA, largely based on data compiled in the MAUDE database. The FDA issued a Public Health Notification in 2008 (2008 PHN) in response to a perceived increase in adverse events related to TVM use in POP and SUI [2]. According to the notification, it was issued based on over 1,000 reports filed during a three-year period (2005–2007) from nine surgical mesh manufacturers of complications related to surgical mesh devices used to repair POP and SUI.
After review of an additional 2,874 reports filed from 2008–2011, the FDA released a related July 2011 safety communication titled “UPDATE on Serious Complications Associated with Transvaginal Placement of Surgical Mesh for Pelvic Organ Prolapse,” which specifically addressed safety concerns related to POP procedures [3]. In response to the UPDATE, The Pelvic Surgeons Network (PSN) endorsed an article published in November 2011 in the International Urogynecology Journal, which refuted certain claims made by the FDA in the UPDATE [4]. They reached dramatically different conclusions in their analysis of the literature cited by the FDA, and asserted that some of the statements and recommendations made in the UPDATE were biased and misleading.
Following submission of the PSN article, but prior to its publication, the Obstetrics and Gynecology Device Panel of the Medical Devices Advisory Committee recommended that vaginal mesh for POP repair be reclassified from Class II to Class III, which requires that premarket clinical studies evaluate new vaginal mesh for POP repair with a mandated control arm comprised of women undergoing repair without mesh. At the same time, they recommended that existing vaginal mesh products for POP repair require 522 postmarket studies to assess risk/benefit. The FDA acted partially on the committee's recommendations in January 2012, ordering manufacturers to conduct the 522 postmarket studies. As recently as May 2014, the FDA had also proposed to honor the committee's other major recommendation in reclassifying TVM for POP as a high-risk device (Class III) [5].
While also based on the literature review and public comments, the initial impetus for the 2008 PHN, 2011 UPDATE, and subsequent Advisory Committee meeting that resulted in the reclassification of TVM was reporting of adverse events collected in the MAUDE database. The inherent limitations of the current reporting system are not always appreciated, however, when MAUDE datasets are used to critique specific devices across all medical specialties, even though such data has widespread implications on care pathways and economics for manufacturers, payers, providers, and patients alike. The objective of this study was to assess the ability of the MAUDE database to objectively capture adverse events for medical devices in the United States using the dataset that inspired the FDA's 2008 PHN for TVM as an example.
Go to : 
MATERIALS AND METHODS
We reviewed 1,103 individual MDRs within the MAUDE database, which were used by the FDA as the basis for the 2008 PHN. These reports were submitted to the FDA from January 1, 2005 to December 31, 2007 and were associated with TVM adverse events in the setting of both SUI and POP.
Two independent reviewers entered the field contents from each individual record into an Excel (Microsoft Corp., Redmond, WA, USA) spreadsheet exactly as they appeared, without editing or interpretation. Any discrepancies were resolved by re-examining the record in question and correcting identifiable errors. Institutional review board/ethics approval did not apply to this study as no human or animal subjects were studied.
Go to :
RESULTS
We analyzed 1,103 MDRs submitted to the MAUDE database from 2005–2007 for pelvic mesh adverse events. Sixteen standardized manufacturers were included (23 unique entries before standardization) representing 83 unique brand entries. Fifty-three unique device types were reported, yet only 33 unique catalog (product) # entries, including 818 (74.2%) that were left blank.
Thirteen unique reporter types spanned all MDRs, including physicians, patients, nurses, risk managers, attorneys, pharmacists, and family members (Table 1). However, 47% of the total submissions did not identify a specific reporter type and included ‘other’ (26%), ‘N/A’ (17%), [blank] (2.4%), ‘unknown’ (1.5%), ‘invalid data’ (<1%), or ‘no information’ (<1%).

Table 1
Types of reporters submitting information to the Manufacturer And User Facility Device Experience (MAUDE) database (n=1,103)
| Reporter types | n (%) |
|---|---|
| Physicians | 519 (47.1) |
| Patients | 25 (2.3) |
| Nurses | 21 (1.9) |
| Risk managers | 9 (0.8) |
| Pharmacists | 5 (0.5) |
| Attorneys | 4 (0.4) |
| Family members | 1 (0.1) |
| Unidentified | 519 (47.1) |
![]()
Report sources included company representatives, user facility representatives or both. Additional sources included other health professionals, attorney and legal representatives, patients/family members/consumers, and unknown/blank/none (Table 2). Notably, 264 of the total 1,103 MDRs were submitted to the FDA by foreign individuals or facilities (not shown).
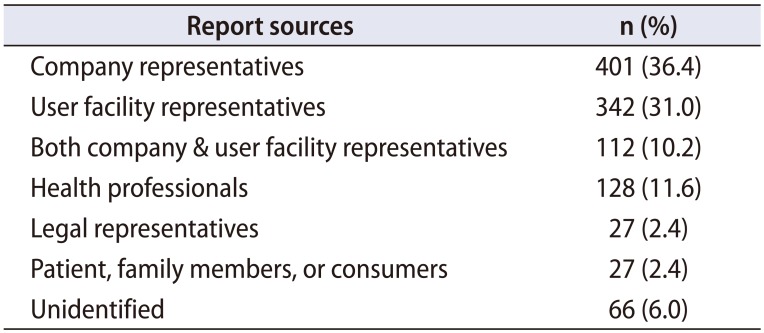
Table 2
Sources of reporters submitting information to the Manufacturer And User Facility Device Experience (MAUDE) database (n=1,103)
![]()
Overall, 91% of submissions cited injury, 7.4% device malfunction, and <1% death with the remainder unspecified (1.2%). When stratified by reporter, 71% of MDRs submitted by nurses cited injury, while 92% of physician submissions reported injury versus device malfunction, other, or death. Patient reporters and attorneys uniformly cited injury (100%). There was also evidence of at least 64 duplicated MDRs, with an additional six reports that sought to represent multiple patients with a single submission. One MDR, in particular, referenced 126 patients, 15 of whom “were identified has [sic] having had possible adverse outcomes…” related to one particular device type.
The overwhelming majority of MDRs cited either a physician or health professional as the device operator, though 138 (12.5%) designated a ‘lay user/patient.’ One thousand sixty-five (1,065, 96.6%) cited at least one adverse event, 316 (28.6%) cited two or more, and 100 (9.1%) reported 3 or more individual adverse events. Importantly, 916 MDRs (83.0%) required intervention or hospitalization with eight reported deaths. Other event outcomes included [blank], ‘other serious,’ ‘other,’ ‘disability or permanent damage,’ and ‘life threatening.’
Finally, at least 28% of brands cited in the analyzed MDRs are no longer on the US market. MDRs featuring products no longer available in the US included: 154 (14.0%) Obtape (Mentor Corp., Minneapolis, MN, USA), 101 (9.2%) Gynecare Prolift (Ethicon, Inc., Somerville, NJ, USA), 31 (2.8%) Obtryx Mesh Sling System (Boston Scientific Corp., Marlborough, MA, USA), 11 (1.0%) Avaulta Plus (C.R. Bard, Inc., Murray Hill, NJ, USA), among others (Table 3).

Table 3
Reported frequency of manufacturers and devices (n=1,103)
![]()
Go to :
DISCUSSION
The recent controversy over the use of TVM in treating women with SUI, and particularly POP, has been well publicized. The FDA's MAUDE database was only a single tool of many used to assess mesh safety and utility in recent years, but it remains as the initial primary signal generator and gatekeeper for thousands of medical devices in use today. There is a clear need for a centralized adverse event reporting system, and the FDA's attempt to fulfill it with the MAUDE database should be applauded. However, our analysis highlights several shortcomings of the current system, which will become ever more visible as healthcare moves into the era of value based care and detailed quality reporting.
By its nature, MAUDE based data cannot be used to accurately estimate true adverse event rates. Although reporting is theoretically mandatory, in practicality it is voluntary, making it impossible to determine the true number of adverse events or the total number of cases performed. Data analysis is further complicated by numerous MDRs seeking to represent adverse events for multiple patients, listing multiple adverse events, and in those with blank or missing data. A comparison on Table 1 to Table 2 is an excellent example of confusing and inconsistent data. While 47.1% of reporters were physicians, it is unclear which of these reporting physicians were the operating surgeons, company representatives, or hospital employees not directly involved in patient care. Moreover, the raw data rarely facilitates knowing whether mesh was placed for POP, SUI, or both. While this may inferred from the type of mesh used, we do not have accompanying patient records to substantiate such inferences. This clearly limits the database's utility for individual analyses.
It is unreasonable to expect uniformity and accuracy when reporters come from varying backgrounds and levels of education and include physicians, nurses, risk managers, lawyers, and patients themselves. The larger issue comes in the form of absent reporter fields, as 47% of all MDRs did not even identify a reporter, though some of these did indicate the less specific ‘Report Source.’ For MDRs listing a reporter, nurses were more likely than physicians to report device malfunction, while patients and lawyers uniformly reported injury. These observations are not necessarily surprising, but they do highlight a difference in motivation between different types of reporters, which may represent another source of bias. One individual may have an interest in either embellishing or withholding the facts of an event, whereas another may do the opposite depending on their relationship to the device and/or patient.
We also note that many pelvic mesh brands cited in MAUDE MDRs are no longer available on the US market. In many cases, it was not possible to identify the exact product cited with absolute certainty, as brand names were often reported incomplete, misspelled, or in generic form (e.g., “Gynecare” versus “Gynecare TVT Secur System”), and often did not include a unique catalog number. Therefore, only MDRs with brand names specific enough to identify them with certainty were counted when determining rates of discontinued products. Thus, the actual rate of MDRs citing discontinued products is likely much higher. It should be noted, however, that while some products were discontinued prior to the 2008 FDA PHN (e.g., Obtape [Mentor Corp.], removed 2006), many were not removed from the market until 2012 or later (e.g., Prolift Pelvic Floor Repair [Ethicon, Inc.]). It is possible that the content and analysis afforded by the MAUDE and resulting PHN and UPDATE contributed to some of these later products being pulled, but the increased scrutiny of many mesh manufacturers in lawsuits also likely played a large role.
Changes in the rates of reported adverse events are not necessarily a reflection of changes in actual adverse event rates. Yet, much of the impetus for the FDA's 2011 UPDATE was simply the increase in MAUDE MDRs filed between 2008 and 2010 [6]. There are many possible explanations for why MAUDE reporting rates may have increased, including the effect of increased publicity and public awareness surrounding a particular family of medical devices. There is also objective evidence to suggest that the use of TVM has increased in general, which might result in increased MDR reports even if AE rates remained constant [7].
Additionally, there has been a major shift in report sources between 2007 and 2011. Forty-three to 53% of the 1,103 MDRs analyzed in this study were sourced from user facilities or other unaffiliated health care professionals while 36%–46% were from device company representatives or distributors. Compare this to the 2,874 MDRs submitted from 2008–2011, of which 94% were sourced from manufacturers, and only five total reports (<0.2%) were submitted by user facilities [8]. Thus, user facilities such as hospitals and surgical centers virtually discontinued submitting reports related to TVM to the MAUDE database after the initial 2008 PHN.
While the MAUDE can be successful in generating a global signal for adverse events, its utility in supplying researchers and policymakers with usable data as a screening tool or for subgroup analyses that may lead to meaningful conclusions regarding the cause(s) of these signals is severely limited. The quality and degree of data reporting by MAUDE users in the case of pelvic mesh was variable and frequently incomplete. Thus the true severity (and even existence) of a problem for a given device is difficult, if not impossible, to define and no reliable denominator of total cases can be determined. These shortcomings are typically overcome with literature review and the initiation of post-market surveillance studies, and improvements to the MAUDE methodology are anticipated with the opening of the American Urogynecologic Society (AUGS) Pelvic Floor Disorders Registry.
There are numerous examples in the literature of data mining techniques aimed at detecting potential signals in the MAUDE database, as well as the counterpart Food and Drug Administration Adverse Event Reporting System (FAERS), which collects information related to medication events [910111213]. Unlike our analysis, these studies often depend on algorithms that target a large number of reports too vast for individual perusal in order to identify numerical correlations between specific adverse events and specific drugs or devices within a class. Acknowledging the limitations of any database, they seek to infer missing data, exclude reports with missing or duplicate data, and do represent strategies that could help overcome some of the limitations described in our analysis [14].
Such post hoc strategies could also be utilized in future registries, though we acknowledge primary collection of complete data would provide superior metrics and generate more robust signals for adverse events. Eliminating barriers for reporting to such registries is of paramount importance, which may include cumbersome user interfaces, reluctance in reporting surgical complications, or simply ignorance that such registries exist. Future versions of electronic medical records may be able to automatically generate reports based on information already recorded by circulators in the operating room. Automatic, compulsory data generation will likely be the only way to truly eliminate missing data in the future, as the potential for human error is ever present.
Recognizing the limitations of the MAUDE database and a need to better track outcomes associated with TVM, the FDA has partnered with industry and professional societies like the American Urological Association, the American Congress of Obstetricians and Gynecologists, and the AUGS, to establish a national mesh registry [15]. Most recently, AUGS announced an initiative to establish the Pelvic Floor Disorders Registry to fill this need [16]. The goals of the registry will be to evaluate the effectiveness, quality of life, and safety associated with surgical and non-surgical treatment options for POP. The registry will also provide a framework for clinical studies to be conducted, including the 95 post-market surveillance studies requested by the FDA from 34 separate manufacturers. Finally, the registry will also allow healthcare providers to track surgeon volume, patient outcomes, and quality measures, and fulfill Centers for Medicare and Medicaid Services, the Physician Quality Reporting System, and maintenance of certification requirements. This ambitious endeavor will serve the interests of those involved in TVM manufacture, use, and regulation, but may also provide a model for future medical devices in the coming era of healthcare safety and quality reporting. In a best case scenario, proactively establishing similar registries for all medical devices may one day preclude the need for post-market surveillance studies altogether, as a comprehensive database capable of tracking a patient from incision to adverse event could provide the wealth of data necessary for fully evaluating a medical device in question. If implemented correctly, such registries may be able to eliminate the problem of missing data by standardizing required responses from users. Such a step would go a long way in mitigating one of the largest flaws of the MAUDE, which is incomplete and missing information.
The principle limitation in this study lies in the nature of the data analyzed. MAUDE MDRs provide mainly categorical and descriptive data, and thus applying sound statistical methods to our analysis was difficult. We quantified and reported categorical variables when possible, and relied on descriptive data to better inform and shape our analysis. At the same time, these limitations reflect the inherent limitations of the MAUDE database, and further argue for the development of alterative reporting systems.
Go to :
CONCLUSIONS
We offer a fresh perspective based on original analysis of one of the major tools used to detect issues related to medical device safety: the MAUDE database. The authors agree with the steps being taken by the FDA and the urogynecologic community to address the safety and efficacy of transvaginal mesh use in POP, and the MAUDE database was successful in inspiring appropriate scrutiny in this case. However, if we are to preserve our patients' welfare, there is a need to improve how we define the safety of a device with a decidedly uncertain safety profile. TVM is not the first, nor will it be the last, medical device category to be scrutinized and be reclassified, but it is important that we understand the tools currently available so that we might improve upon them in the future.
Go to :
 XML Download
XML Download