

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Sacral agenesis is a rare malformation, occurring in approximately 0.005–0.01% of the general population (1). It shows varying degrees of vertebral dysgenesis combined with spinal cord abnormalities. Magnetic resonance imaging (MRI) most accurately demonstrates deformity of the spine and the spinal cord, as well as combined anomalies. These are key factors in determining the treatment plan and also in predicting patient's prognosis.
Traditionally, sacral agenesis is categorized into 4 types according to the degree of sacrum dysgenesis and the articulation between the spine and pelvis (12). Classification is also made based on the morphology of the spinal cord. We describe a case of partially bilateral agenesis of the sacrum (type II), and club-shaped (chisel-shaped) spinal cord disruption, with a review of the literature.
CASE REPORT
A 4-year-old girl was brought for further evaluation of lifelong urinary incontinence and hesitancy. She was born at 37 weeks 4 days with a body weight of 3.3 kg. Neonatal hyperbilirubinemia was noted at birth, which was treated with phototherapy for one week. Recurrent acute pyelonephritis and constipation had been a problem throughout her life, requiring numerous hospitalization for conservative management. After consultations with various physicians, she was diagnosed as having "emotional incontinence" and neurogenic bladder, and was started on medical therapy.
She is the only child of the family; both parents were nonconsanguinous and in good health. There was no other history of known congenital anomalies or inheritable diseases in either parent's family, in particular, maternal diabetes mellitus.
On physical examination, she was able to walk unassisted, although she was easily tired. No evidence of other orthopedic abnormalities was noted in both lower extremities. A number of studies were performed to evaluate for urinary incontinence, including abdominal ultrasound, dimercaptosuccinic acid scan and voiding cystourethrogram. Both kidneys showed overall normal morphology without functional abnormalities, but the diagnosis of neurogenic bladder was confirmed.
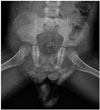
The anteroposterior view of a plain pelvis radiograph (Fig. 1) revealed first and second sacral elements with an absence of other lower sacrum and coccyx bilaterally. Both iliac bones were articulated with S1.
L-spine MRI (Fig. 2) showed hypoplastic S2 to be the lowest vertebra, with bilaterally symmetric agenesis of the distal sacrum. Articulation of both iliac bones was noted at the S1 level. The cord was terminated above the L1 level. The caudal end of the spinal cord was club-shaped (chisel-shaped) with blunted and angulated margins. Separation of the anterior and posterior spinal roots of the cauda equina was also observed. Neither abnormal signal intensities of the spinal cord, such as syringohydromyelia, nor spinal canal stenosis, were present. Distended urinary bladder with a coarse trabeculation suggested that the neurogenic bladder was not accompanied by other urogenital abnormalities, such as renal dysplasia, hydronephrosis, or anorectal malformation. The final diagnosis was type II sacral agenesis with mild urological dysfunction.
DISCUSSION
Sacral agenesis, also known as caudal regression syndrome, is characterized by caudal vertebral agenesis or dysgenesis, including hemisacral anomalies, most often in combination with spinal cord malformations (3). The incidence of this syndrome is very rare, approximately 1 in 7500 births, regardless of gender (4). The etiology of the sacral agenesis is not fully understood. According to other reports, it is postulated that the development of the caudal portion of the spine and spinal cord during the 28th gestational week is disrupted (56). Others have suggested prenatal exposure to various teratogenic substances (i.e., lithium and sulfamides) as causative agents (6). The relationship between sacral agenesis and genetic problems (such as HLXB9 mutation) has also been debated (7). To date, the only factor reported to be significantly associated with sacral agenesis is maternal diabetes (1). Sacral agenesis occurs in about 1 in 100 infants born to diabetic mothers (1), representing an increase of nearly 200 times the incidence observed in the general population.
Renshaw (2) has classified sacral agenesis into 4 types, according to the severity of vertebral agenesis and the articulation between the remaining vertebrae and iliac bone. Partial agenesis of the sacrum is further classified as unilateral asymmetric (type I) form, or bilateral symmetric (type II) form. In cases of total sacral agenesis (with or without partial lumbar agenesis), type III involves the articulation of both iliac bones with the lowest lumbar vertebra, whereas in type IV, both iliac bones are fused posteriorly along the midline. Bilateral symmetric partial agenesis of sacrum below S2, with iliac bone articulation normally at S1 was noted in this case, which is consistent with type II sacral agenesis.
Various levels of the vertebral column termination have been observed in the literature, from T8–9, to only involving the coccyx (8). In most cases, 1 or 2 vertebra distal to the lowest intact vertebrae are hypoplastic. Furthermore, Barkovich et al. (8) reported that the thoracolumbar spine is normal above the agenetic segment in only 65% of patients; the remaining had combined anomalies, such as vertebral body fusion, hypoplastic spinous process, etc.
Evaluation of combined spinal cord abnormalities is also necessary, and MRI is the most preferred diagnostic modality of choice. The 2 characteristic shapes of the spinal cord terminus were reported; blunted or wedge-shaped (type I) and tethered spinal cord (type II) (48). The wedge-shaped cord terminus is more common, showing the dorsal aspect of the cord extending more caudally than the ventral aspect. In addition, an abnormal course of the spinal roots of the cauda equina, that is, separation of the anterior and posterior spinal roots, can be seen, termed a "double bundle shape" (4). On the other hand, type II shows low-lying, tapered, caudal spinal cord tethered by tight filum or other combined congenital anomalies (lipoma, lipomyelomeningocele, or terminal myelocystocele). In this case, the typical morphological features of type I spinal cord abnormality were observed, including 2 separate cauda equina and blunted conus medullaris.
The level of the cord terminus and vertebral agenesis was consistent with neurological manifestations (8). However, several reports have demonstrated sensory function to remain lower than the level of vertebral regression; therefore, sensory function test is not a reliable tool for evaluation of the regression level (48). Also, type I spinal cord abnormality is known to be more frequently associated with urinary bladder dysfunction and stable neurological defect, whereas type II may demonstrate progression of neurological symptoms, which are often surgically correctable (4).
Sacral agenesis can be a part of other systemic syndromes: OEIS (omphalocele, cloacal exstophy, imperforate anus and spinal deformities) and Currarino triad (partial sacral agenesis, anorectal malformation and presacral mass) (49). Moreover, combined anomalies in the pelvic region are not uncommon. In the genitourinary system, renal agenesis, dysplasia and/or ectopia may be associated with sacral agenesis. In cases with anorectal malformations, anatomic details and involved level of anorecal atresia should be investigated. MRI is the preferred method of choice for the diagnosis of sacral agenesis for superior demonstration of spine and spinal cord deformity, as well as clearer depiction of other combined anomalies.
In conclusion, the exploration and diagnosis of sacral agenesis is important in providing appropriate management. Management depends on the types of sacral agenesis (categorized by the affected spine and spinal cord morphology) and combined anomalies (mild life-long urological symptoms to severe gastrointestinal and musculoskeletal disabilities). Here we have reported MRI findings of partially bilateral agenesis of the sacrum (type II) and club-shaped (chisel-shaped) spinal cord disruption in a child.

 XML Download
XML Download