

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Paraganglioma is a rare neuroendocrine tumor of extra-adrenal chromaffin cell origin. The tumor can occur in any region within the confines of normal paraganglion cells, typically in sites such as the para-aortic region at the level of renal hila, the organ of Zuckerkandl, the thoracic para-spinal region, bladder, and head and neck. The tumor is most commonly observed in the second or third decade of life, and it has no known gender-related predilection. Patients with paraganglioma are less likely to present with hormonal symptoms compared to those with adrenal pheochromocytoma (1). Few case reports of primary hepatic paraganglioma have been published because of its rarity. We describe the case of a 52-year-old man with pathologically confirmed primary hepatic paraganglioma, which showed a predominately hemorrhagic septated cystic mass, and peripheral marked enhancement of the solid portion with persistent enhancement.
CASE REPORT
A 52-year-old man was referred to our hospital for further evaluation of an incidental hepatic mass that was detected on computed tomography (CT) scan during evaluation of left-sided flank pain. The patient's medical history was unremarkable, with the exception of hypertension, which was controlled by medication. The patient had no clinical signs or symptom at presentation, and laboratory test results were within the normal range. The hepatitis virus serological test results and the alpha-fetoprotein levels were also normal.
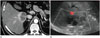
An outside contrast enhanced CT scan revealed a large septated cystic mass in liver segment 6 with a peripheral solid portion that showed avid enhancement (Fig. 1). Both adrenal glands were nonspecific. The initial impression was an atypical complicated cyst or an abscess. Ultrasonography revealed a septated cystic mass with a peripheral hypoechoic rim. Vascularity at the septum and peripheral rim was not evident in a Doppler study (Fig. 1).
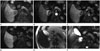
On a follow-up magnetic resonance imaging (MRI) scan after 2 months, the mass showed no significant interval change. In addition, the lesion was observed to be a primarily cystic mass with a peripheral solid component that showed high signal intensity on T2-weighted imaging, and low signal intensity on T1-weighted imaging. The solid portion enhanced markedly during the arterial phase of enhanced MRI with gadoxetic acid, and portal venous and delayed phases of enhanced MRI revealed slightly high signal intensity compared with the surrounding liver parenchyma. The solid portion showed low signal intensity during the hepatobiliary phase. The cystic portion also showed fluid-fluid levels that were indicative of hemorrhage. A hypervascular tumor, such as hepatocellular and neuroendocrine carcinomas, was suspected based on imaging characteristics (Fig. 2).
An ultrasound-guided biopsy was performed, and a pleomorphic myxoid neoplasm, possibly malignancy was confirmed. Subsequently, a right hepatectomy was performed. Gross analysis showed a well-demarcated cystic mass with internal septation, including hemorrhage in the cystic components. Microscopic examination revealed large polygonal cells with distinctive basophilic cytoplasm. Immunohistochemical analysis of the resected tumor revealed that the tumor was positive for chromogranin A, synaptophysin, and S-100, and electron microscopic examinations indicated multiple neurosecretory granules, suggestive of paraganglioma (Fig. 3). It was concluded that the tumor was non-functioning paraganglioma because the patient had no specific symptoms and evidence of excessive catecholamine secretion, except for hypertension, which was readily controlled by oral medication.
No other abnormality was indicated by 2-(fluorine 18) fluoro-2-deoxy-d-glucose positron emission tomography (PET) following right hepatectomy. In a follow-up enhanced CT scan after 2 years, no evidence of tumor recurrence was found.
DISCUSSION
Paraganglioma is a rare neuroendocrine tumor of extra-adrenal chromaffin cell origin, and it can arise from any paraganglia present within the body (2). In general, neural cells are not present in the normal liver, which makes primary hepatic paraganglioma extremely rare. There have been few case reports of hepatic paraganglioma. The underlying cause of primary hepatic paraganglioma is related to ectopic chromaffin tissues (3).
One of the most important factors in the treatment of primary hepatic paraganglioma is elimination of the possibility of metastasis from pheochromocytoma, or paraganglioma at other sites (4). The patient in the present case had undergone several imaging modalities, including pre-surgical CT, MRI, post-surgical PET-CT and follow-up CT, to eliminate the possibility of pheochromocytoma or paraganglioma at other sites; however, there were no other abnormal findings. Hormonally active paraganglioma may present with symptoms such as palpitation, headache, and high blood pressure, which may facilitate the diagnosis with the indication for metaiodobenzylguanidine (MIBG) scintigraphy. The sensitivity of MIBG scintigraphy is 81%, which may render this method susceptible to false negative results (5). In the present case, we did not perform MIBG scintigraphy since the patient did not present with any characteristic symptoms or signs of catecholamine excess.
Hepatic paraganglioma exhibits similar radiologic features to those of pheochromocytoma in the adrenal glands. Pheochromocytoma typically displays avid contrast enhancement due to a rich capillary network, and it may show delayed washout, and internal hemorrhage, cystic changes, necrosis, and internal calcifications are frequently reported (6). Hepatic paraganglioma displays avid contrast enhancement due to a rich capillary network in the arterial phase of post-enhancement scans, like pheochromocytoma. Cystic areas that occur as a result of hemorrhage, necrosis, or heterogeneous enhancement, may be observed in enhanced CT scans. In MRI studies, the tumor shows low to iso signal intensity compared with normal liver parenchyma, and high or heterogeneous signal intensity on T1-weighted, and T2-weighted images, respectively. T1 and T2 signal intensity may vary depending on the histologic variation (7).
A few case reports have demonstrated that hepatic paraganglioma displays marked enhancement, with a poorly enhanced central portion on enhanced CT, mimicking fibrolamellar hepatocellular carcinoma (HCC); however, there are no reports about the cystic hemorrhagic changes in the tumor, as seen in the present case. Wang et al. (8) reported high grade hepatic neuroendocrine carcinoma with peripheral enhancement and internal necrosis with evidence of hemorrhage, as was observed in the current case. It is difficult to distinguish hepatic paraganglioma from other hypervascular liver tumors such as HCC and hepatic neuroendocrine tumors, through the use of imaging alone. HCC typically exhibits arterial hyper-enhancement, with a portal or delayed wash-out appearance. HCC does not usually show delayed persistent enhancement or fluid-fluid levels, such as those seen in the current case. Furthermore, HCC is rarely observed in populations that do not have any preceding risk factors such as liver cirrhosis and chronic hepatitis. Hepatic neuroendocrine tumors have similar imaging findings to those of hepatic paragangliomas, and they also display marked arterial enhancement, delayed persistent enhancement, and hemorrhagic, cystic changes (8).
It is considered impossible to make a diagnosis of primary hepatic paraganglioma solely through radiologic measures due to its rarity and nonspecific radiologic characteristics (9).
A limitation of the present case was the lack of assessment of the serum or urine catecholamine levels following hepatectomy. However, this was due to the fact that the patient's blood pressure remained unchanged after surgery.
In conclusion, we report an extremely rare case of primary non-functioning hepatic paraganglioma that was successfully treated by hepatectomy. The tumor was revealed to predominantly consist of a cystic mass resulting from hemorrhage with marked enhancement of the solid portion. The present case shows that hepatic paraganglioma may be considered in the differential diagnosis in cases of hemorrhagic well-enhanced hepatic masses.

 XML Download
XML Download