

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Inflammatory myofibroblastic tumor (IMT), also known as inflammatory pseudotumor, is a rare and benign process with myofibroblastic spindle cell proliferation. IMT usually displays an indolent course but its radiologic appearance can mimic malignancy. Although it can occur in any organ, the lung is the most commonly involved (1). Pulmonary IMT typically manifests as a single, peripheral, and sharply circumscribed mass with lower lobe predominance (2).
Here, we present a case of pulmonary IMT with atypical radiologic and clinical manifestations which was treated successfully with prednisone. To our knowledge, there has not been a case report on multifocal pulmonary IMT rapidly progressing into masses in young adults.
CASE REPORT
A 27-year-old woman with a one-week history of dry cough, fever, and blood-tinged sputum was admitted for evaluation of resting dyspnea which had started a few hours prior to admission. The patient reported night sweating and weight loss of 3 kg over one month.
Initial laboratory investigations revealed a white blood cell count of 15.7 K/mcL, erythrocyte sedimentation rate of 54 mm/hr (0–22 mm/hr) and C-reactive protein level of 13.6 mg/L (0–10 mg/L), consistent with an inflammatory reaction.
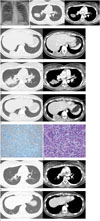
The initial chest radiograph on admission revealed a round mass in the right lung field and some faint mass-like densities in the right infradiaphragmatic and left upper retrocardiac areas (Fig. 1A). Subsequent CT scans of the chest revealed multiple, sharply-circumscribed, round necrotic masses in the middle and lower lung fields (Fig. 1B, C). Within one month, the pulmonary masses progressed into larger ill-defined masses with newly developed small pleural effusion (Fig. 1D, E).
The patient was initially treated with antibiotics (Penicillin G and clindamycin) for two months without clinical and radiologic improvement. She underwent fine needle biopsy of the largest mass in the superior segment of right lower lobe. This revealed inflammatory myofibroblastic cells consistent with IMT and gave negative Ziehl-Neelsen stains for granuloma (Fig. 1F, G). Immunohistochemistry of the tissues revealed positive markers for actin, vimentin and oncogenic anaplastic lymphoma kinase (ALK-1) reflecting the myofibroblastic and neoplastic nature of the lesion. The patient was then treated with prednisone (30 mg per day for 1 month then gradually decreased down to 10 mg per day) for 6 months. The pulmonary masses and clinical symptoms showed complete resolution without recurrence for over a year (Fig. 1H, I).
DISCUSSION
The IMT is a rare benign tumor without known etiology which occurs more commonly in young age groups. It can involve various areas of soft tissues, with pulmonary involvement being the most prevalent (1). IMTs are histologically composed of myofibroblastic cells and may mimic malignant processes by recurrence and infiltration into nearby tissues (34), posing a challenge to radiologists in differentiating them from malignant lesions. Therefore, radiologic and histopathologic correlation is essential for diagnosis.
Most pulmonary IMTs appear on radiographs as a single lesion but their slow progression differentiates them from lung cancer. Typical radiologic features of pulmonary IMT include being a solitary and peripheral mass, which exists as a sharply circumscribed mass located predominantly in the lower lobes (2). Because of the rather non-specific and variable radiological findings, there are many differential diagnoses for pulmonary IMTs including malignant lesions such as primary lung carcinoma and pulmonary metastasis, and benign tumors such as granuloma, hamartoma, and hemangioma (25). In our case, the initial radiological differential diagnosis included bacterial infection, fungal infection, actinomyocosis, and even pulmonary peripheral T cell lymphoma considering the rapid progression of the lesion. Pathologic differential diagnoses were eosinophilic granuloma, fungal infection and focal organizing pneumonia.
To our knowledge, pulmonary IMT's initial manifestation as multifocal masses, as seen in this case, is relatively unusual. Sometimes it can manifest as multifocal nodules especially in children where IMTs with rapid recurrence have been reported occasionally (6). Pulmonary IMT is known to occur in multiples in 5% of total cases (2). There was a case report on recurrent multiple nodular lesions which were diagnosed as pulmonary IMT (7), but the patient did not initially present with multiple masses.
Coffin et al. (8) found that about half of the IMTs have a clonal cytogenetic aberration which is involved in activation of the ALK-receptor tyrosine kinase gene on the short arm of chromosome 2 at 2p23. Their study reported that ALK reactivity may be a favorable prognostic indicator, and also more likely to be associated with the atypical characteristics of IMTs. The pathological and immunohistochemical findings in this case revealed positive reactivity for actin, vimentin, and ALK, consistent with the myofibroblastic and neoplastic nature of the lesions (3). In this case, the initial pulmonary nodule progressed into ill-defined mass-like lesions, which showed complete resolution with six months of corticosteroid treatment. There has been no evidence of recurrence seen in clinical and radiologic follow-ups for one year. Positive ALK expression could explain the favorable outcome despite the atypical manifestation in this case.
A previous case report categorized 32 cases of IMT into three types–organizing pneumonia, fibrous histiocytoma, lymphoplasmacytic type–using a histopathological spectrum where organizing pneumonia type is distinct in its gradual resolution of intraalveolar exudates (9). Based on the chronic inflammatory cells found in the tissue and the complete resolution of pulmonary masses, it is possible that the pulmonary IMT in this case could be categorized as organizing pneumonia.
Regarding treatment of pulmonary IMT, surgery is the treatment of choice at the present (8). However, corticosteroids, non-steroidal anti-inflammatory drugs and chemotherapy have also been discussed as proven treatments (8). Corticosteroid treatment alone for six months had a dramatic therapeutic effect on the patient in this case.
IMT is still disputed in its characteristics, and it involves multiple organs without a known etiology. Due to its unpredictable nature and tendency to recur, careful attention during
 XML Download
XML Download