

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
An inflammatory pseudotumor (IPT) is a rare tumor composed of proliferative myofibroblasts, fibroblasts, histiocytes, plasma cells, and lymphocytes (1). IPT is mainly seen in young patients and often involves the lung and orbit, but it may occur in virtually any anatomic location and in patients of any age.
Although IPT usually presents as an isolated mass in a single organ, it sometimes manifests in the form of multifocal lesions that are concentrated in one anatomic region (2). Few IPT cases have been reported in the ureter and bladder. The lesions are usually benign, but some abdominal cases have exhibited malignant potential or metastasis. Recently, some experts have suggested classifying IPT into three categories, including plasma cell-rich IPT [also known as lymphoplasmacytic or immunoglobulin G4 (IgG4)-related IPT] (3). In this case report, we describe the clinical, imaging, and pathology lab results of an IgG4-related IPT that occurred in a patient's periureteral area, paravesical space, and prostate.
CASE REPORT
In month, year, a 49-year-old man came to our urology department complaining of voiding difficulty during the previous two months. Laboratory test results and urine analysis were not remarkable. The patient underwent contrast-enhanced abdominal and pelvic CT, transuretheral resection of the prostate (TURP), and transurethral incision of the prostate. Based on pathology test results, he was diagnosed with benign prostatic hyperplasia with prostatitis. After three days of using a foley catheter, his symptoms resolved and he was discharged.
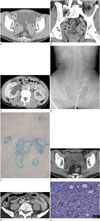
One year later, the patient presented again with right flank pain and a palpable mass on his right abdominal wall. The mass had been present for two months. As before, the results of the initial laboratory tests and urine analysis were not remarkable. Axial contrast-enhanced CT imaging revealed the presence of three newly developed and ill-defined heterogeneous, delayed, enhancing masses located in the bilateral periureteral areas and right paravesical space. The bilateral periureteral masses had encased the distal ureters, and the left periureteral mass abutted the inferior surface of the bladder (Fig. 1A). There was also evidence of infiltration of the bilateral obturator muscles (Fig. 1A).
A coronal reformatted contrast-enhanced CT image showed irregular wall thickening and luminal narrowing of the bilateral distal ureters (Fig. 1B). An axial contrast-enhanced CT image showed bilateral hydroureteronephrosis, which was due to distal ureteral narrowing caused by the periureteral masses (Fig. 1C). A retrograde pyelography image showed hydronephrosis in the right kidney with double-J catheter insertion in the right ureter, and segmental dilatation with stenosis in the left distal ureter (Fig. 1D).
We attempted a sono-guided biopsy of the right paravesical mass in order to confirm our diagnosis from the pathology test results. Ultrasound showed a heterogeneous hypoechoic mass containing feeding vessels (not shown). We were unable to penetrate the tumor with a biopsy needle, so we obtained only a small amount of the specimen for testing. Pathology results revealed chronic inflammation with fibrosis. Surgical biopsy was then done in order to acquire additional tissue. During the operation, the right paravesical mass was removed, but the bilateral periureteral masses were not completely removed because of difficulties presented by severe adhesion. The right paravesical mass and both periureteral masses contained grayish yellow fibrotic tissue with a firm consistency. Cut surfaces showed grayish, firm, rubbery tissue mixed with fat tissue. No necrosis was apparent.
Microscopic examination of the paravesical and periureteral masses revealed diffuse infiltration of spindle cells with severe fibrosis, and also showed marked infiltration of plasma cells, lymphocytes, histiocytes, and eosinophils (Fig. 1E). Immunohistochemical (IHC) staining of the spindle cells was positive for smooth muscle actin (SMA) and CD34, but negative for desmin. The SMA-positive spindle cells suggested myofibroblastic differentiation. These histologic findings were compatible with IPT. In addition, the prostate tissue that had been obtained the previous year via TURP showed multiple plasma cells and was compatible with the diagnosis of IPT upon retrospective review.
After surgery, the patient received radiation therapy and steroid hormone therapy for treatment of residual masses and consequent hydroureteronephrosis. A follow-up CT scan (Fig. 1F) indicated that the bilateral hydroureteronephrosis and eccentric ureteral wall thickening had substantially improved, and the bilateral periureteral masses were reduced in size. After three mo-nths, another CT scan revealed a soft tissue lesion encasing the left proximal ureter (Fig. 1G). Because of the relapsing pattern of lesions encasing the organ and growing awareness of IgG4-related sclerosing disease among clinicians, the pathologist performed additional IHC staining to identify suspected IgG4-related IPT. The specimen contained a large volume of IgG4-positive plasma cells (Fig. 1H) and CD68-positive histiocytes. We could not calculate the ratio of IgG4-positive/IgG-positive plasma cells because IgG IHC stain was not available in our hospital. There were no histopathological findings suggestive of malignancy, infection, or granulomatous diseases. A later serologic test performed in an outpatient clinic showed that the patient's serum IgG4 level was within the normal range (2.46 mg/dL, conventional normal range, 135 mg/dL or less).
The patient continued to receive steroid treatment. Serial follow-up CT scans showed that the thickening of the left distal ureteral wall had resolved, and four years after surgery there were no recurrent or new mass-like lesions found in the abdomen or pelvis.
DISCUSSION
IPT is a rare entity of unknown etiology (1245). Tumors tend to occur more frequently in young patients, but there is nevertheless a wide age distribution. IPT does not occur more often in one gender or the other. The clinical onset of IPT may be slow or rapid, and patients may initially present with symptoms of chronic inflammatory illness (6). IPT can affect any organ or anatomic site, although the urogenital tract is generally not involved (45). Few cases of IPT have been reported that involve the bladder. Cases in the ureter or kidney are even more rare; these latter tumors usually manifested as single benign lesions in patients presenting with hematuria and/or abdominal pain (45). Our patient was one of the rare cases of multi-organ IPT that affected the periureteral area, paravesical space, and prostate. He presented with right flank pain and a palpable right abdominal wall mass.
Although imaging studies of IPT yield findings that are nonspecific, contrast-enhanced CT or MRI tests usually show delayed enhancement from the large amount of fibrous components in these tumors. Our case was similar. IPT of the ureter has been described as a submucosal polypoid mass or an infiltrative lesion, but we are aware of only five reports of infiltrative mass lesions in the ureter (36). In our case, it was not possible to distinguish whether the origin of the masses near the bilateral distal ureters was the ureter or the periureteral area. Additionally, multi-organ IPT has been associated with irregularity of the ureters, multifocal enhancing wall thickening at the adjacent right and inferior surfaces of the bladder, and infiltration in the left pelvic floor muscle, based on CT imaging.
Examination of pathology lab results generally reveals myofibroblastic differentiation in the fibroblastic spindle cells and variable staining with myoid markers including SMA, muscle-specific actin, and desmin (7). However, a large amount of tissue is needed to accurately identify IPT via pathology lab testing, and needle biopsies are not feasible for this purpose. Therefore, it is often difficult to make a precise diagnosis before surgery (6).Diagnosis is usually made at the time of surgical intervention and later confirmed by examination of pathology test results and IHC staining, combined with clinical and radiologic findings (6).
Recently, some pathologists have suggested that IPT can be divided into three categories: cellular IPT [inflammatory myofibroblastic tumor (IMT)], plasma cell-rich IPT (lymphoplasmacytic or IgG4-related IPT), and fibrohistiocytic IPT. The pathogenesis of each type is different. Fibrohistiocytic IPT may result from an inflammatory process with prominent fibrosis and marked histiocytic and myofibroblastic proliferation (3). Conversely, IMT may be caused by a neoplastic process, while IgG4-related IPT may result from an autoimmune process.
Specific diagnostic criteria for IgG4-related IPT have not yet been established, but Yamamoto et al. (8) have reported that bland-looking spindle cell proliferation with fibrosis and inflammatory infiltrate of lymphocytes and plasma cells, are the common morphologic features in both IMT and IgG4-related disease. In their report (8), the number of IgG4-positive plasma cells and the ratio of IgG4-positive/IgG-positive plasma cells were both significantly lower in IMT than in IgG4-related disease [mean 6.4/high power field (HPF) vs. 178.3/HPF (p < 0.0001), 3.0% vs. 67.5% (p < 0.0001), respectively]. They therefore suggest that IgG4 does not play an important role in the pathogenesis of IMT. Deshpande et al. (9) proposed diagnostic categories of IgG4-related disease based on pathology test results; the category they call "histologically highly suggestive of IgG4-related disease" requires the presence of at least two of the following histological features: dense lymphoplasmacystic infiltrate; fibrosis, usually storiform in character; and obliterative phlebitis.
In our patient's pathology lab results and IHC staining, the paravesical and periureteral masses revealed diffuse infiltration of spindle cells, severe fibrosis, prominent plasma cells, lymphocytes, histiocytes, eosinophils with abundant IgG4-positive plasma cells, and SMA-positive spindle cells, all of which suggest a diagnosis of IgG4-related IPT. The patient's postoperative serum IgG4 level was within the normal limits. An elevated serum IgG4 level is not required for diagnosis of IgG4-related IPT, although it may be helpful for identifying some cases (10).
It is important to identify IPT type before selecting a treatment. Although surgery remains the preferred treatment for IPT, other alternative such as radiation therapy and steroid therapy have been associated with successful regression of IPT (1). The prognosis for IPT is usually good, and most tumors do not recur after complete surgical excision (16). Complete resection, however, can be difficult, especially in the abdomen; these tumors may have a greater potential for malignancy or metastasis, and are associated with an overall mortality rate up to 7% (15). Patients with IgG4-related IPT may prefer alternative or additional treatments such as steroid based therapy rather than radiation therapy or surgery (7). Since our patient was initially diagnosed with general IPT, he underwent surgery first and was then treated with radiation and steroid therapy. Although his multi-organ IPT responded to radiation and steroid therapy after surgery, his condition was later aggravated and he required additional treatment a year later.
In conclusion, we have described the case of a 49-year-old man with rare multi-organ IgG4-related IPT involving the periureteric area, paravesical space, and prostate and presented the results of his CT imaging, pathology lab testing, and IHC staining. When numerous plasma cells are present in a patient with IPT, a physician should consider a diagnosis of IgG4-related IPT and look for signs of autoimmune disease before selecting a tre-atment.
 XML Download
XML Download