

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Acute hyperammonemic encephalopathy results from hyperammonemia associated with acute liver failure. Acute hyperammonemic encephalopathy can cause sudden altered mentality or even abrupt progression to coma state (1, 2). Because acute hepatic encephalopathy is typically a clinical diagnosis, imaging studies were rarely performed until recently.
There is a specific pattern of imaging findings on acute hyperammonemic encephalopathy. According to recent studies, the most common involving sites of acute hyperammonemic encephalopathy are insula, diffuse cerebral cortex, cingulate cortices, and bilateral thalami (1, 2). Additional sites involved are subcortical white matter, basal ganglia, and brainstem. However, they are not common sites of involvement. MRI findings can give clues for diagnosis of acute hyperammonemic encephalopathy (2, 3).
Acute hyperammonemic encephalopathy can mimic hypoxic-ischemic encephalopathy (HIE) because both involve diffuse cerebral cortex and bilateral thalami. However, in the setting of acute elevation of ammonia levels, radiologists should consider possibility of acute hyperammonemic encephalopathy (1, 2, 4).
CASE REPORTS
Case 1
A 56-year-old male patient presented nausea, vomiting and lethargy. He had past medical history of liver cirrhosis due to chronic hepatitis B. Two weeks ago, he started anti-tuberculosis treatment with isoniazid, ethambutol, rifampicin, and pyrazinamide due to pulmonary tuberculosis. Laboratory testing showed ammonia level of 218 µmol/L (normal range: 0-34 µmol/L). Liver function tests (LFTs) showed levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) at 239 U/L and 300 U/L, respectively. Total bilirubin (TB) was 25.23 mg/dL. His pulmonary tuberculosis medication was stopped because of hepatotoxicity of isoniazid, rifampicin, and pyrazinamide.
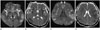
By hospital day 1, MRI was performed due to his decreased mentality. On fluid-attenuated inversion-recovery (FLAIR) images, there was no significant abnormality. But diffusion-weighted imaging (DWI) showed symmetrical bilateral increased signal intensities at the insula, both diffuse at temporo-fronto-parieto-occipial cortices, cingulate cortex, bilateral dorsomedial thalami. Apparent diffusion coefficient (ADC) maps confirmed reduced diffusion (Fig. 1).
After 2 days, multi-organ-failure was developed. Conservative treatment and continuous renal replacement therapy was performed. But by hospital day 6, he died from multi-organ failure.
Case 2
A 47-year-old male patient visited the emergency department with episodes of confusion. Upon arrival, the vital signs of patient were stable with saturation level of oxygen in hemoglobin (SaO2) of 100%. He had flapping tremor on neurologic examination.
Laboratory testing showed ammonia level of 169 µmol/L (normal range: 0-34 µmol/L). Hepatitis A virus (HAV) antigen and HAV immunoglobulin M were positive, implying acute hepatitis A. LFTs showed the levels of AST and ALT at 1755 U/L and 5539 U/L, respectively. TB was 9.83 mg/dL.
DWI was performed for his abruptly decreased mentality. On FLAIR images, there were no significant abnormalities. DWI and ADC map showed diffusion restriction at the insula, diffuse cerebral cortex, cingulate cortex, and dorsomedial thalami similar to first case (Fig. 2).
The patient's hyperammonemia was treated with lactulose enema and conservative treatment. By hospital day 2, his mental status progressed to stupor and he was transferred to other hospital for liver transplantation because of fulminant hepatitis.
DISCUSSION
Symptoms of patients with acute hyperammonemic encephalopathy include sudden onset of drowsiness and seizures. Untreated hyperammonemia can lead to permanent brain injury. Therefore, early diagnosis and prompt treatment of hyperammonemia is crucial to prevent long-term sequelae and to avoid fatal complication such as brain edema, herniation, even death (1, 2, 5).
Ammonia is produced in the gastrointestinal tract as a by-product of protein digestion and bacterial metabolism. It is metabolized in the liver as urea through the urea cycle (2). When the metabolic capacity of the liver is overwhelmed, elimination depends on the kidneys, skeletal muscle, and even brain (1, 2, 6). In the brain, ammonia affects the excitatory glutaminergic N-methyl-D-aspartate receptors and gamma-aminobutyric acid receptors. This pathway makes subsequent cell swelling and even apoptosis (2, 6).
The most common cause of hyperammonemic encephalopathy is acute liver failure. Other etiologies are portosystemic shunt surgery, drugs (valproic acid, barbiturates, narcotics, alcohol, and chemotherapy), gastrointestinal bleeding, urinary tract infection with urease-producing organism, ureterosigmoidostomy, parenteral nutrition, bone marrow transplantation, solid organ transplantation, severe muscle exertion, septic shock, and congenital inborn errors of metabolism (2, 3, 7).
MRI findings of chronic liver disease are well known to show bilateral hyperintensities of the globus pallidus, subthalamic region, and midbrain on T1-weighted images (1, 8). Radiologic findings of acute hyperammonemic encephalopathy are less well reported. However, with increased using of DWI, distinct MRI changes of acute hepatic encephalopathy have been reported recently (1, 2, 4).
U-King-Im et al. (2) presented 4 adult patients with acute hyperammonemic encephalopathy with restricted diffusion involving the insula, cingulate cortex and diffuse cerebral cortex. Arnold et al. (9) also reported a case of a diffuse cortical necrosis secondary to hyperammonemia. This report showed restricted diffusion at diffuse cerebral cortices, insula, and the cingulate cortex similarly. According to previously reported literature about acute hyperammonemic encephalopathy of children, bilateral involvement of the insular cortex and cingulate gyrus were strikingly common features in pediatric patients (10). It is unclear why the insula and cingulate coretex are particularly susceptible to toxic effects of ammonia (1).
Choi et al. (4) reported severe case of cortical laminal necrosis after hyperammonemic encephalopathy. In this case, involved lesion was predominant in the watershed zones or the parietooccipital regions, suggesting that hemodynamic factors might be involved in the disease.
Involvement of brain regions other than the insula or cingulate cortex, diffuse cerebral cortices are more variable. Other areas are deep gray matter such as bilateral thalami, basal ganglia, subcortical white matter, periventricular white matter, and brainstem. Involvement of these sites are rarely observed, except bilateral thalami (1, 2). McKinney et al. (3) found out that thalamic involvement was present in 85% of 20 patients on FLAIR and in 70% of 20 patients on DWI. They reported that thalamic involvement was very common, even more frequent than cerebral cortex involvement.
Rosario et al. (1) and McKinney et al. (3) have reported that if the involved regions are thalami, dorsomedial thalami are frequently affected. Our two cases showed increased signal intensity in bilateral dorsomedial thalami on DWI. Dorsomedial thalami lesions are also known as predominant site of Korsakoff's syndrome. Korsakoff's syndrome results from conditions of malnutrition and vitamin deficiency due to chronic alcoholism. Chronic alcoholic patients have a tendency to hyperammonemia due to repeated liver damage. We suggest that lesions of dorsomedial thalami in Korsakoff's syndrome might be associated with patient's previous subclinic hyperammonemic encephalopathy (6).
According to McKinney et al. (3), plasma ammonia levels correspond well to the extent of MRI abnormality. Plasma ammonia level also correlates with clinical outcome. It was suggested that MRI features only moderately correlated with clinical outcome (3). Therefore, radiologists should consider clinical setting, especially plasma ammonia level (3).
Early vague clinical manifestations of hyperammonemia such as anorexia, lethargy, disorientation can be seen with lower plasma ammonia levels of 60 µmol/L. Early MR imaging changes can be seen at such lower levels. U-King-Im et al. (2) reported patent MRI image with ammonia levels of 55 µmol/L (normal range: 0-34 µmol/L). The patient in that case had extensive MR imaging changes and made an excellent recovery without significant neurologic deficit with proper treatment.
Acute hyperammonemic encephalopathy can cause severe long-tern sequelae, such as intellectual disability or even death. However, if aggressive treatment is instituted, cortical changes in hyperammonemic encephalopathy can be potentially reversible (1, 2, 5).
Acute hyperammonemic encephalopathy may be misinterpreted as HIE due to their similar predominantly involving sites. HIE involves diffuse cerebral cortex and thalamus similarly. Arnold et al. (9) regarded acute hyperammonemic encephalopathy as HIE initially. However, with combination of plasma ammonia level and absence of hypoxic event, they could make early diagnosis of acute hyperammonemic encephalopathy (1). It is very important to make differential diagnosis. Because acute hyperammonemic encephalopathy is reversible if early diagnosis and proper treatment is done. However, HIE is irreversible. Other predominantly involving sites of HIE are bilateral basal ganglia and hippocampus. Knowing other additional predominantly involving sites of HIE and considering patient's medical history, laboratory findings can make precise diagnosis of acute hyperammonemic encephalopathy.
In summary, we suggest that the following four main predominant sites are involved in acute hyperammonemic encephalopathy: insula, cingulate cortex, diffuse cerebral cortex, and dorsomedial bilateral thalami. It is important to make early diagnosis of acute hyperammonemic encephalopathy as well as differential diagnosis, because acute hyperammonemic encephalopathy is a reversible and very fatal condition.

 XML Download
XML Download