

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Proteus syndrome (PS) was described as a discrete clinical entity by Cohen and Hayden (1) in 1979. In 1983, the term Proteus syndrome was coined by Wiedemann et al. (2) after the polymorphous Greek god Proteus who has the ability to change his appearance. PS is known to be caused by a mosaic, activating AKT1 mutation (c.49G > A, p.Glu17Lys). The mutation has been identified in only 2 of 38 peripheral blood DNA samples, so a blood test is a poor way to establish the diagnosis. Lindhurst et al. (3) suggest that the clinical criteria for PS alone may be sufficient for the diagnosis without identifying the mutation, except in early cases when the clinical findings are not definitive. The purpose of this report is to familiarize the reader with multiple clinical and radiologic manifestations of PS including skeletal, soft-tissue and visceral anomalies as well as tumors.
CASE REPORT
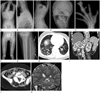
A 14-year-old girl was referred for the evaluation of her scoliosis. She underwent an excision for the hyperostosis of her external auditory meatus in another hospital. The thoracolumbar hump on her back was revealed in an Adam's forward banding test. It is the positive sign for the presence of a thoracolumbar scoliosis. On physical examination, she had a long, asymmetric face with a relative enlargement of the right side, an uneven macrodactyly affecting both hands and a tortuous, serpentine, skin-bulging superficial lesion with an overlying skin discoloration, in her left lower leg, suggesting a vascular malformation. The patient did not have any neurological symptom. Laboratory studies revealed no abnormality. There was no family history of congenital anomaly. Radiographs were obtained of the vertebrae, skull, hands, feet and extremities. And then, MR imaging were also performed of the whole spine, unenhanced CT of the chest, enhanced CT of the abdomen-pelvis and MR imaging of the brain for further evaluation. Spine radiographs showed an asymmetric overgrowth of the multiple vertebrae with severe resultant scoliosis, abnormal anteroposterior spinal alignment and posterior scalloping of the thoracolumbar vertebra (Fig. 1A-C). On MR imaging and CT, the axis appeared as megaspondylodysplasia with a right sided hyperostosis, leading to a marked reduction in the patient's mobility. Skull radiographs revealed asymmetric multifocal calvarial thickening with increased convolutional changes and an asymmetric overgrowth of the right sided mandible and facial bone (Fig. 1D). Hands radiograph showed an uneven macrodactyly of the third and fourth right digit and of the first and second left digit; a clinodactyly of the second and fourth right digit and of the second left digit; diffuse soft tissues thickening in the second and third right rays and in the second left ray; and an associated hyperostosis and calcified soft tissue mass in the third right ray (Fig. 1E). There was no demonstrable abnormal finding on feet radiographs. Radiographs of the lower extremities showed a leg length discrepancy with the right femur slightly longer than the left and a bowing in the left femur; disproportionate overgrowths of the tibiae to the fibulae; and a superficial tortuous soft tissue density in her left calf, suggesting a vascular malformation (Fig. 1F, G). An unenhanced CT of the chest revealed small cystic changes in both lower lungs, with the right side more severe than the left as well as an asymmetrical fat deposition in the posterior chest wall (Fig. 1H). An enhanced abdominal-pelvic CT revealed splenomegaly, increased retroperitoneal fat and asymmetric fat infiltrations in the paraspinal muscles (Fig. 1I, J). The brain MR imaging revealed a slight enlargement of the right cerebral hemisphere with cortical thickening and a mainly in the parietal lobe diminished sulcation and an ipsilateral ventricular enlargement. These findings were consistent with the diagnosis of a right-sided asymmetric megalencephaly (Fig. 1K).
DISCUSSION
PS is a rare congenital hamartomatous condition that produces multifocal overgrowth of tissue derived from any of the three germinal layers (4). This disorder is characterized by progressive mosaic overgrowth of skin, bones, muscles, fatty tissues and blood and lymphatic vessels as well as by visceromegaly, lung cysts and a predisposition to pulmonary embolism. The most common causes of a premature death in Proteus syndrome are pulmonary embolism and respiratory failure. Predisposing factors for pulmonary embolism in these patients include vascular malformations, surgical convalescence and a very restricted mobility in severe cases of Proteus syndrome (5). The syndrome is rarely associated with benign or malignant tumors. PS is caused by a somatic activating mutation in AKT1 (c.49G > A, p.Glu17Lys). Nevertheless, a knowledge of the multiple specific manifestations of PS is necessary for clinical and imaging diagnosis (3). Biesecker et al. (6) and Jamis-Dow et al. (7) set up diagnostic criteria in 1999. There are general and specific criteria to diagnose a patient with PS. The general mandatory criteria are mosaic distribution of the lesions, progressive course and sporadic occurrence. Mosaic or random distribution is the hallmark of PS and it means that only some body parts show signs of overgrowth while others are unaffected. This disease commonly progresses in childhood rapidly, but may slow down or stabilize during early adolescence. Patients of PS have no family history of congenital anomaly that presents similar features of overgrowth. Specific criteria or category signs are also necessary and should include either one sign from category A or two from category B or three from category C. The single sign in category A, cerebriform connective tissue nevus, appears to be pathognomonic for the diagnosis. But, it is not a common finding in patients with PS. It is most frequently found as a gyriform gross thickening of the soles. The category B includes linear epidermal nevus, disproportionate overgrowth and specific tumors, such as bilateral ovarian cystadenoma and parotid monomorphic adenoma before or during the second decade of life. The signs of the category C are dysregulated adipose tissue, vascular malformations, lung cysts and an abnormal facial phenotype. A pulmonary disease may not be rare in PS, although it has not been widely recognized. It may include pulmonary venous dilatation and bullous disease. Small lung cysts in PS, as seen in this case, can progressively and irreversibly change into an extensive bullous disease (8). On chest radiographs the pulmonary disease in PS can be misdiagnosed as chronic or interstitial pneumonia and can be easily overlooked if the thorax is deformed, in particular, when severe thoracic scoliosis is present (8). In the present case, the chest radiograph showed non-specific findings and small lung cysts were depicted on chest CT. In spite of the high radiation risk in our young patient, a CT was inevitable because of the limitation of ultrasound study for an abdomino-pelvic evaluation with sound beam attenuation due to the patient's subcutaneous, intrapelvic and retroperitoneal fat hypertrophies. Progressive skeletal abnormalities such as macrodactyly, scoliosis, asymmetric overgrowths and limb length discrepancy are the most frequent and striking findings in patients with PS, followed by soft-tissue abnormalities such as fatty, muscular and vascular malformations. The hyperostosis of external auditory meatus, asymmetric megalencephaly and splenomegaly are rare visceral involvements that were seen in this patient. Asymmetric megalencephaly or hemimegalencephaly is a rare congenital abnormality produced by a hamartomatous overgrowth of all parts or a part of the cerebral hemisphere. In addition to hemimegalencephaly, brain radiological features in a patient with PS may include callosal dysgenesis, neuronal migration disorder and calcification. Epilepsy and epileptic syndrome may also be associated (9). Our patient didn't present with a clinical history of seizure. The diagnosis of PS in this case was made with the condition that the patient satisfied all of the mandatory general criteria and specific criteria from both, category B (disproportionate overgrowth of limbs, vertebrae, skull, external auditory canal, spleen and cranium) and category C (dysregulated adipose tissue, vascular malformation and cystic lung changes).
The differential diagnosis should include other overgrowth syndromes. The disorders most commonly confused with PS are Klippel-Trenaunay syndrome, neurofibromatosis type 1, epidermal nevus syndrome and hemihyperplasia-multiple lipomatosis syndrome (6, 7, 10). The tissue overgrowth in Klippel-Trenaunay syndrome is usually secondary to cutaneous capillary or venous vascular malformations, whereas the overgrowth of bone and other tissues may occur independently of vascular malformations in PS. In PS, the limb overgrowth usually is absent or mild at birth, whereas in Klippel-Trenaunay syndrome it is present at birth and is commonly severe (6, 7, 10). The osseous overgrowths were not associated with vascular malformations in the present case. In neurofibromatosis type 1, massive enlargement of the skin and soft tissues may occur, and they often associated with plexiform neurofibromas and a proliferation of nerve sheath fibromyxoid tissue. The resultant mass effect and pressure-induced changes may deform the underlying bone. Neurofibromatosis type 1 affects first-degree relatives because of its autosomal dominant inheritance and it is characterized by cafe-au-lait spots, cutaneous and subcutaneous tumors, axillary or inguinal freckles, Lisch nodules, two or more neurofibroma or one flexiform neurofibroma, sphenoid dysplasia or typical long bone abnormalities such as pseudoarthrosis and optic glioma. Neurofibromatosis type 1 was an unlikely diagnosis in our case due to the absence of those signs (6, 7, 10). Epidermal nevus syndrome is a rare sporadic neurocutaneous disorder characterized by an epidermal nevus and cerebral anomalies. Central nervous system anomalies are typically severe and ipsilateral to the nevus. The spectrum of central nervous system abnormalities includes those ranging from ventriculomegaly to severe migrational disorders. No epidermal nevus was shown in the patient of the present case (10). Hemihyperplasia-multiple lipomatosis syndrome, a very rare sporadic disorder with mosaic distribution, is characterized by an asymmetric overgrowth over half of the body and multiple lipomata, resulting in an asymmetry between the right and the left body sides. Cutaneous capillary malformations may occur in some instances. Mild to moderate signs are present at birth. Progressive overgrowth is more marked in PS, whereas hemihyperplasia-multiple lipomatosis syndrome is relatively stable (10).
In conclusion, PS is a complex disorder with multisystem involvement and great clinical variability. Diagnosis of this disease will depend entirely on the clinical and imaging features until a specific mutation is identified. Knowledge of the multiple highly characteristic radiologic manifestations of Proteus syndrome and an appropriate use of imaging modalities are therefore essential for an accurate diagnosis of this condition.
 XML Download
XML Download