

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Granular cell tumor (GCT) of the neurohypophysis, also referred to as choristoma, myoblastoma, or granular cell myoblastoma, is a rare neoplasm of the neurohypophysis. The 2007 revision of the World Health Organisation (WHO) Classification of Tumors of the Central Nervous System has unified the various histopathologically similar tumor entities arising from the suprasellar or sellar regions under the GCT of the neurohypophysis (WHO Grade I). The WHO 2007 definition of a GCT is as follows: "An intrasellar and/or suprasellar mass arising from the neurohypophysis or infundibulum, composed of nests of large cells with granular, eosinophilic cytoplasm due to abundant intracytoplasmic lysosomes". Previous reports described GCTs as well-defined intrasellar or suprasellar masses visible on magnetic resonance imaging (MRI), showing an hypointense signal or an isointense signal on T1, T2-weighted images (1234) with calcification being rarely seen as evident in only 3 of the 47 cases reported thus far (12). In this article, we present a symptomatic case of GCT of the neurohypophysis with an emphasis on the magnetic resonance (MR) signal intensity characteristics.
CASE REPORT
A 60-year-old female with a 3-month history of reduced visual acuity in her right eye was referred for evaluation. She complained of a visual field defect in the temporal half of her right eye for a period of 3 months and an continuous headache for a period of 15 days. Contrast-enhanced MR and CT imaging were performed.
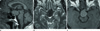
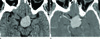
MRI revealed a large (3.2 × 2.5 × 1.7 cm), well defined, smooth marginated mass in the pituitary gland with loss of normal posterior lobe hyperintensity. The evaluation of MR images revealed an isointense signal on a T1-weighted image (Fig. 1A), and a hypointense signal on a T2-weighted image (Fig. 1B). After contrast administration, the neoplasm showed mild contrast enhancement (Fig. 1C). Stenosis of the distal portion of left internal carotid artery and compression of right midbrain by the mass lesion were noted. CT scans showed the tumor density and the attenuation patterns. On the nonenhanced CT scan, the tumor was homogenous and showed a slightly higher density (about 53 HU) (Fig. 2A), and on the enhanced CT scan, the tumor showed a homogenous contrast enhancement (Fig. 2B).
Subtotal tumor resection was performed via the supra-or456bital craniotomy approach. During the operation, when the sellar floor was opened up, an abnormal yellowish demarcated mass was detected at the midline of the sellar and the suprasellar areas. The mass was compressing the superomedial aspect of the left optic nerve and was enclosing the left distal internal carotid artery. Internal decompression of the left optic nerve and a subtotal tumor resection was performed. A histolopathological examination showed sheets of polygonal cells with abundant eosinophilic granular cytoplasm (Fig. 3A). The cytoplasmic granules were positive for a periodic acid-Schiff stain (Fig. 3B), and resistant to diastase digestion. Immunohistochemical staining for S-100 protein (Fig. 3C) showed positivity in the cytoplasm and the nucleus of tumor cells. Tumor cells were negative for glial fibrillary acidic protein (GFAP), neurofilament, and cytokeratin. The Ki-67 labelling index is less than 1% (Fig. 3D). Postoperatively, the patient suffered from transient diabetes insipidus, but the patient's right eye visual acuity improved.
DISCUSSION
The most common tumors of the neurohypophysis are the GCTs (1). GCTs are a relatively common incidental autopsy finding seen in up to 17% of random adult autopsy results (4), but reports of such symptomatic cases are rare (2). These tumors are clinically silent until they cause symptoms due to their size. As a consequence, they are usually large in size by the time they are diagnosed. The most common clinical presentation includes neurological symptoms such as visual field defects and headaches, and endocrinopathies such as hypopituitarism or hyperprolactinemia. Patients usually present in the fifth decade of life, and women are more commonly affected than men (2 : 1 ratio).
Etiology and histogenesis of GCTs of the neurohypophysis are uncertain, and this uncertainty about their cellular origin (4) has led to GCTs being described under many names such as choristoma, myoblastoma, and granular cell myoblastoma. Some investigators have argued that GCTs of the Central Nervous System originate from the Schwann cells, but the others have suggested that the cell of origin for the neurohypophyseal GCTs is the pituicyte, which is a modified astrocyte (1). These tumors are composed of large polygonal cells with a small nuclei and granular cytoplasm (3), and can be extremely vascular. Hence, careful preoperative planning is crucial. The transphenoidal surgical approach, which is the therapy of choice in most sellar benign symptomatic tumors (1), can be risky in the patients with GCT as they present a high risk of bleeding.
These tumors are composed of closely approximated, large, round or oval cells lacking any characteristic arrangement. The most typical feature of these tumors is an abundance of small granules with a rich protein content in the cytoplasm (5). In this case, the tumor showed a hypointense signal on T2-weighted image and a slightly high density on the precontrast enhanced CT scan. This is probably due to the rich protein content within the intracytoplasmic granules. Although these imaging findings are not in accordance with the previous reports, they can be considered as the characteristic imaging findings in granular cell tumors.
The immunohistochemical findings varied, and some researchers have reported a positive nuclear reaction for S-100 protein (6). Similarly, variable findings have been reported for GFAP.
A few rare cases of GCT of the neurohypophysis have been reported in the radiology literature (37). The MRI characteristics reported are nonspecific, and show a well-defined intra- and suprasellar mass. The anatomical site of the mass can be of help in the diagnosis of GCT when the mass is located posteriorly within the pituitary gland, and the normal posterior lobe hyperintensity cannot be identified. Our case also showed a large mass in the pituitary gland with loss of normal posterior lobe hyperintensity. However, adenoma still continues to be the most common tumor of the neurohypophysis.
Also due to the rarity of GCT lesions, no "typical" signal intensity pattern has been described. In this case, the MRI signal intensities are inconsistent with the findings reported by the other investigators. In this case, the tumor showed an isointense signal on T1-weighted image and a hypointense signal on a T2-weighted image with mild contrast enhancement. In the previous five case reports, the tumor had shown an isointense signal on both the T1- and T2-weighted images with strong homogenous contrast enhancement (234). Calcification within the tumor was reported only in three patients and no calcification was seen in the other case reports including this one (689). It appears that the presence of calcification might be related to the size of the tumor.
According to the previous reports, GCT MRI findings are similar to meningioma and are indistinguishable from adenoma, the most common pituitary tumor.
The natural history of GCTs is still uncertain. Although Schaller et al. (1) have commented on the tumor's slow growth and its benign histology, paradoxically, they have also cited the relatively a short survival period (2-26 months) in the patients treated conservatively. The survival period in the patients treated with surgery alone (42 ± 52 months) or in the patients treated by radiotherapy (81 ± 91 months) was distinctly longer. Clinical examination usually reveals bitemporal hemianopsia, optic nerve atrophy, and reduction in the growth rate of body hair in the male patients (1347).
Often, only a subtotal tumor resection is performed due to the tumor's toughness, vascularity, and its proximity to the hypothalamus. In some cases, the pituitary stalk had to be sacrificed in order to achieve a complete tumor resection (8). In addition, transient postoperative diabetes insipidus is very common. The role of postoperative radiotherapy is controversial. While Becker and Wilson (9) thought in 1981 that radiotherapy would be ineffective, Wilson in 1992 recommended radiotherapy in cases of incompletely resected tumors (8). Of the 46 GCT patients reported in the literature reviews, five patients died during the postoperative period and two died after the second operation for tumor recurrence (6).
In conclusion, the following findings of: a radiological diagnosis of an intra and or a suprasellar mass, variable signal intensities on T1- and T2-weighted images, and variable enhancement after contrast administration, a lack of identification of the normal posterior lobe hyperintensity, and an anatomical location within the posterior part of the hypophysis, may be suggestive of a diagnosis of GCT. A hypointense signal on a T2-weighted MR image is in accordance with the rich protein content of intracytoplasmic granules and can be considered as a signal intensity characteristic of GCT for neurohypophysis.

 XML Download
XML Download