

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Hypophysitis, an inflammation of the pituitary gland, is clinically and radiologically important because it mimics tumor-like lesions in the sellar area and may produce mass effects. Xanthomatous hypophysitis is a type of primary hypophysitis with a very low prevalence.
In previously reported cases (123), patients with xanthomatous hypophysitis sometimes presented with a headache, sexual dysfunction, and/or diabetes insipidus. A pituitary lesion with cystic change is evident by imaging as well as the diminishment of testosterone, FSH (follicle-stimulating hormone), and LH (luteinizing hormone) levels.
The authors report a case of xanthomatous hypophysitis, which presented as an intrasellar cystic lesion, hypogonadism, and hyperprolactinemia. The report also includes a literature review.
Case Report
A 40-year-old man was admitted with a 2-month history of a gradually aggravated visual disturbance and a headache history of several years. There was no familial history of trauma or autoimmune disease. A physical exam revealed a limitation in the right lateral visual field with normal extraocular movements.
Initial laboratory testing revealed anterior pituitary dysfunction and a reduction in testosterone (<0.01 ng/mL), FSH (1.2 mIU/mL), and LH (0.3 mlU/mL) levels; hyperprolactinemia (36 ng/mL) was also reported (Table 1). Other laboratory findings were within normal ranges and electrocardiography (ECG) demonstrated a normal sinus rhythm.
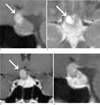
An initial brain CT showed normal brain parenchyma, but a sellar mass was suspected. Magnetic resonance imaging (MRI) showed diffuse enlargement of the pituitary gland with a lobulated small cystic portion (about 9 × 13 mm in size) at the suprasellar region. The cystic portion was hyperintense based on T1- and T2-weighted images (Fig. 1), and was suspected to be lipid, a high protein content region, or hemorrhage (4). The enlarged gland was homogenously enhanced (like normal pituitary gland tissue) and the adjacent meninges and showed no enhancement or thickening (Fig. 1). However, the pituitary stalk was mildly thickened. The right portion of the optic chiasm was compressed by the sellar lesion, especially by its cystic portion. The adjacent cavernous sinus and basal cistern showed no abnormal findings.
Surgery was performed using a trans-sphenoidal approach. The pituitary gland was found to be enlarged and the cystic lesion was visualized. As the cystic portion was being resected, it leaked a yellowish fluid. The operation was completed, leaving the rest of the intrasellar lesion. Histologically, hematoxylin and eosin stained sections showed pituitary gland tissue with surrounding xanthoma cells and multinucleated giant cells containing cholesterol clefts. CD68 (DAP staining) was positive, confirming the presence of xanthoma cells. Furthermore, there was no evidence of a pituitary adenoma or craniopharyngioma (Fig. 2). After surgery, methylprednisolone was continuously administered on a daily basis. A week later after surgery, the visual disturbance had markedly improved, FSH was slightly increased (2.4 mIU/mL), and prolactin slightly decreased (22.9 ng/mL). However, testosterone levels were still depressed. The patient remains on desmopressin acetate therapy because of symptoms indicating diabetes insipidus development. A follow-up MRI performed 3 months after surgery demonstrated the removal of the cystic portion, but the gland enlargement and mild stalk thickening persisted (Fig. 1C).
Discussion
Hypophysitis is an inflammation of the pituitary gland by caused by a bacterial, viral, or fungal infection in an immunocompromised background. In addition, systemic disorders, such as sarcoidosis (5), Wegener's granulomatosis, Langerhans histiocytosis, Takayasu's disease, Crohn's disease, and Rosai-Dorfman syndrome (6), may account for secondary hypophysitis. Primary hypophysitis is classified as lymphocytic, granulomatous, or xanthomatous (2), based on its clinical and radiologic features (12).
Lymphocytic hypophysitis predominantly occurs in female patients, has solid features, and consists of many lymphocytes and plasma cells microscopically. Patients with lymphocytic hypophysitis have ACTH deficiency, but diabetes insipidus is uncommon.
Granulomatous hypophysitis shows no sexual predilection, and has mixed cystic and solid imaging features. Unlike lymphocytic hypophysitis, diabetes insipidus (DI) is common in granulomatous hypophysitis, and its prognosis is often fatal.
Xanthomatous hypophysitis, the least common type of primary hypophysitis, was determined to be a separate disease entity by Folkerth et al. in 1998 (3), who described its unique histological finding as a foamy histiocyte infiltration into the adenohypophysis. Small lymphocytes and liquefactive cyst formation are characteristic features of xanthomatous hypophysitis. In addition, the presence of multinucleated giant cells has been suggested to be a distinguishing feature of Xanthomatous hypophysitis.
Cheung et al. (2) reported a 32-year-old xanthomatous hypophysitis patient who presented with oligomenorrhea and hyperprolactinemia, 3 years after pregnancy, and also compared the lymphocytic and granulomatous appearance of xanthomatous hypophysitis. Most reports on the subject state that xanthomatous hypophysitis may have a more cystic appearance than other hypophysitis cases. Furthermore, cystic lesions were reported to contain a yellowish fluid, which was also evident with our case.
Xanthogranulomatous inflammations are sometimes referred to as cholesterol granulomas and are also found in the middle ear, mastoid, paranasal sinuses, and choroid plexus (7). Xanthoma cells are rarely found in primary or secondary pituitary diseases such as xanthomatous hypophysitis, xanthogranulomatous hypophysitis, or xanthogranuloma, in the sellar area or diseases such as Erdheim-Chester disease, adamantinomatous craniopharyngioma, or Rathke's cleft cyst. Paulus et al. (7) compared 37 xanthogranulomatous lesions and 59 classical adamantinomatous craniopharyngioma lesions, and managed to differentiate the two using patient age, site, and prognosis. Xanthogranulomas in the sella area occur at a younger age, are limited to the sella, and have a better prognosis than craniopharyngioma.
Hypopituitarism is a characteristic finding in primary hypophysitis, which explains the common complaint of sexual dysfunction, and the greater awareness (due to greater prevalence) in lymphocytic hypophysitis than in the other two disease forms. It has been suggested that this anterior pituitary hormone insufficiency may be due to the inflammatory destruction of pituitary acinar cells (8). Adrenal deficiency and hypothyroidism are uncommon in xanthomatous hypophysitis. Posterior pituitary disorders, such as, prolactinemia, are considered to be caused by diffuse inflammatory infiltration rather than by compression of the stalk or posterior lobe of the pituitary gland.
In our case, the cystic lesion was hyperintense according to T1-weighted imaging. This finding is important but nonspecific because it is also present in pituitary adenomas, Rathke's cleft cysts, craniopharyngiomas, abscesses, mucoceles, and dermoid cysts (4).
Furthermore, our patient presented with hypogonadism, hyperprolactinemia, and postoperative diabetes insipidus. Hypogonadism is a common finding in primary hypophysitis, and is present in all xanthomatous hypophysitis cases. Other pituitary lesions, including pituitary adenoma, do not appear to cause reductions in sex hormone levels. However, hyperprolactinemia, commonly seen in pituitary microadenoma, is an exception,
On the other hand, patients with all types of hypophysitis may have hyperprolactinemia, but it is not a common feature (<30%). Our patient's hyperprolactinemia persisted after operation, which was probably due to a xanthomatous type that was unresponsive to steroid therapy. Growth hormone deficiency is a common feature in xanthomatous hypophysitis, despite its absence in our case. In addition, our patient had a visual disturbance, which is unusual, and has not been previously reported in xanthomatous hypophysitis. On the other hand, visual disturbances are common in lymphocytic types. In our case, diabetes insipidus developed postoperatively. This is not in-line with 25~50% of xanthomatous cases reported, which had diabetes insipidus before surgery DI (9).
The recommended medical treatment for hypophysitis is glucocorticoids, but its effectiveness is often inadequate and patients may even be nonresponsive - especially due to xanthomatous lesions. Therefore, surgery is recommended and usually has a good prognosis. However, it is not effective in all xanthomatous patients. For example, hypogonadism and hyperprolactinemia were not improved in our case.
Xanthomatous hypophysitis is an uncommon pituitary disorder that presents with pituitary gland enlargement, hypogonadism, and hyperprolactinemia. If a cystic portion is depicted by MRI, and the pituitary gland is enlarged with accompanying symptoms of hypogonadism, and hyperprolactinemia xanthomatous hypophysitis, should be considered the first line differential diagnosis.


 XML Download
XML Download