

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Uterovaginal duplication with an obstructed hemivagina is a rare disease entity that is often accompanied by ipsilateral renal agenesis. It was recognized as early as 1922 (1) and is sometimes referred to as Herlyn-Werner-Wunderlich (HWW) syndrome (23). The association of renal agenesis with ipsilateral blind hemivagina was reported as Herlyn-Werner syndrome in 1971 (4), while the association of renal aplasia, bicornuate uterus with isolated hematocervix, and a simple vagina was reported by Wunderlich in 1976 (5). This complex of anomalies is observed in adolescents and young women with progressive dysmenorrhea, abnormal pain, menstrual irregularities and a pelvic mass. Early and accurate diagnosis of this entity is important, and a resection of the obstructing vaginal septum can provide pain relief and prevent further complications (67).
We report the initial and follow-up imaging findings of a case of HWW syndrome associated with thin glomerular basement membrane disease of the contralateral kidney, which to our knowledge is the first radiological report in the domestic literature.
Case Report
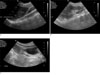
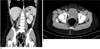
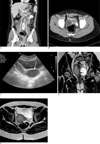
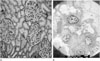
A 12-year-old female visited the pediatric department with the chief complaint of left flank pain and fever for a 1 week duration. Her previous history revealed renal agenesis, which was diagnosed at age of 31 months. Since then she had multiple episodes of urinary tract infections. At age of 9 years, she was hospitalized for another episode of urinary tract infection, at which ultrasonography revealed right renal agenesis and a very hypoplastic uterus with indiscernible detailed structures (Fig. 1). Ultrasonography and contrast enhanced CT scan at age of 11, revealed right renal agenesis (Fig. 2A) and a newly developed cystic lesion in the right pelvic cavity (Fig. 2B). Seven months later, she was admitted for fever and left flank pain. Upon admission, urinalysis revealed red blood cells (4+, 20-29/HPF) with white blood cells (5-10/HPF). The contrast enhanced CT scan (Figs. 3A, B), ultrasonography (Fig. 3C) and MR images (Figs. 3D, E) after admission revealed the absence of her right kidney, compensatory hypertrophy with multifocal scarring in the left kidney, and a further enlarged cystic lesion in the pelvic cavity, which was eventually confirmed to be right hematocolpos in uterine didelphys. A percutaneous core needle biopsy of the left kidney was performed to confirm the cause of the microscopic hematuria, with a subsequent pathological diagnosis of thin glomerular basement membrane disease (Fig. 4).
DISCUSSION
The precise etiology and pathogenesis of HWW syndrome is still unknown. It has been considered to represent anomalous Mullerian (paramesonephric) and Wolffian (mesonephric) duct development (89). The internal genital organs and lower urinary tract are derived from two paired urogenital structures that develop in both genders; the Wolffian ducts and the Mullerian ducts. In females, the Mullerian ducts are located just lateral to the Wolffian ducts, which act as inductor elements, grow downward and toward the midline, crossing the Wolffian ducts, coming in contact with each other, and fusing to form the uterovaginal canal, from which the fallopian tubes, uterus and upper two-thirds of the vagina develop. A range of uterine anomalies can occur as a consequence of the non-development or failure of fusion of the distal segments of the Mullerian ducts, such as hypoplasia/agenesis, unicornuate, didelphys, bicornuate, septate and arcuate uterus. Wolffian ducts are the origin of the kidneys, and inductor elements for adequate Mullerian duct fusion. Therefore, a developmental anomaly of the caudal portion of one of the Wolffian ducts may be the cause of the unilateral renal agenesis associated with imperforate hemivagina (610).
Renal agenesis is a fairly common congenital anomaly with an unknown definite etiology. Renal agenesis may be unilateral or bilateral. Bilateral renal agenesis is a rare anomaly that is incompatible with life, occurring in only one or two per 10.000 births.
Renal agenesis occurs when there is: (1) an absence of the metanephric blastema; (2) ureteral bud maldevelopment; or (3) lack of induction of the metanephric blastema by the ureteral bud. Sometimes a solitary kidney is the result of post-natal involution of a multicystic dysplastic kidney and a hydronephrotic kidney.
Associated ipsilateral urogenital anomalies are common, and include an absence of the vas deferens, unicornuate uterus and an absence or cysts of the seminal vesicle. Other associated anomalies include skeletal abnormalities, anorectal malformations, cryptorchism and cardiovascular abnormalities. The classical association is observed in VATER syndrome, where developmental lesions may include vertebral and ventricular septal anomalies, anorectal atresia, tracheal and esophageal lesions and radial bone abnormalities (11).
HWW syndrome is usually discovered at puberty, shortly after menarche due to the cyclic, increasing low abdominal pain secondary to hematocolpos resulting from longstanding, retained, partially clotted menstrual blood in the obstructed hemivagina.
The syndrome remains unrecognized at first because the menstrual flow from the unobstructed hemivagina is patent. Hematocolpos is suspected only months after menarche and the diagnosis is generally made only if this syndrome is suspected (39). A right-sided prevalence of an obstructed hemivagina has been described (38).
Another Mullerian ductal anomaly that can be associated with a renal anomaly is Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, which is characterized by congenital aplasia of the uterus and the upper part (2/3) of the vagina in women showing normal development of their secondary sexual characteristics and a normal 46XX karyotype. It may be isolated but it is associated more frequently with renal, vertebral, and to a lesser extent, auditory and cardiac defects (12).
Early recognition and prompt surgical removal of the obstructing vaginal septum with drainage is the definitive treatment (3913) that will rapidly relieve the symptoms and prevent complications related to chronic cryptomenorrhea, such as endometriosis, pelvic adhesion and infectious conditions of pyocolpos, pyometros and pyosalpinx (313). Resolution of the obstruction is also essential for preventing secondary endometrisosis (1415). Fertility is not decreased significantly in women with a didelphic uterus (6).
Along with IgA nephropathy, thin basement membrane disease (also known as benign familial hematuria and thin basement membrane nephropathy) is the most common cause of asymptomatic hematuria. Thinning of the basement membrane of the glomeruli in the kidneys is the only abnormal finding with this disease. Its importance lies in the fact that it has a benign prognosis, and maintains a normal kidney function. This condition has been reported to be associated with Alport syndrome or loin pain hematuria syndrome (16171819). However, to our knowledge, this is the first report of this condition being associated with HWW syndrome.
In conclusion, when the absence of a kidney is found in an infant or young child, the small size and tubular shape of the uterus can make it difficult to evaluate the combined uterine anomaly. Therefore, the patient should be followed-up until the end of puberty because an appropriate preoperative diagnosis and treatment will prevent unnecessary procedures and offer the relief of symptoms. Furthermore, in HWW syndrome patients, it is necessary to consider the TGBM of the contralateral kidney when microscopic hematuria is present.
 XML Download
XML Download