

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a medical condition characterized by the accumulation of fat in the liver. NAFLD is used to describe a range of states from simple accumulation of triglycerides (TG) in hepatocytes to the condition with inflammation, fibrosis, and cirrhosis. The prevalence of NAFLD has been reported in the range of 10% to 24% of normal and 58% to 74% of obese people, depending on the population studied and the analytic methods used [12]. The simple accumulation of TG in hepatocytes is not considered a serious disease condition and is often self-limited; however, 4% to 27% of the patients with severe fatty liver that is not treated properly could develop cirrhosis, which can ultimately develop into hepatocellular carcinoma [3].
Various drugs, hormones, and multiple genetic defects in energy metabolism can cause fat accumulation in the liver. However, the most common cause of NAFLD is insulin resistance, which is closely related with obesity, diabetes, and dyslipidemia [45]. Changing lifestyles including weight loss, diet, and exercise to control the related conditions such as obesity and diabetes is the treatment of choice for fatty liver diseases. Drugs that improve insulin resistance and dyslipidemia, antioxidants, and bile acids are currently used for treatments, but the effects are limited, and some of the drugs often produce serious side effects. This review will discuss molecular changes that mediate hepatic TG accumulation in insulin resistance and focus on recent animal studies that suggest a new therapeutic target for hepatic steatosis and hypertriglyceridemia.
SOURCES OF HEPATIC TRIGLYCERIDES
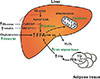
The fatty acids for hepatic TG are derived from three sources: (1) dietary fat, (2) free fatty acids released from adipose tissue, and (3) hepatic de novo lipogenesis. A series of metabolic alterations occur in the liver in insulin-resistant states. In normal people, 80% of hepatic fatty acids are derived from adipose tissue, while those from de novo lipogenesis and diet account for 5% and 15%, respectively. However, in obese and hyperinsulinemic NAFLD patients, the portion from de novo lipogenesis is increased to 26% [6]. In livers of rodent models of insulin resistance, increased rates of fatty acid synthesis are a significant contributor of the development of hepatic steatosis [7]. In the setting of increased lipogenesis, mitochondrial fatty acid oxidation is reduced. In adipocytes, hormone-sensitive lipase activity is increased in insulin resistance, which results in continuous release of free fatty acid through TG lipolysis. Therefore, in an insulin-resistant state, the increased influx of fatty acids from adipocytes, increased de novo lipogenesis, and reduced fatty acid oxidation result in TG accumulation in the liver (Fig. 1).
REGULATION OF DE NOVO LIPOGENESIS BY SREBP TRANSCRIPTION FACTORS
Hepatic de novo lipogenesis is mainly regulated by transcriptional control of the genes involved in the process by transcription factors such as sterol regulatory element binding protein-1c (SREBP-1c), carbohydrate response element binding protein (ChREBP), liver X receptor α, and peroxisome proliferator-activated receptor γ [8910]. Among these, SREBP-1c is the main transcription factor that regulates hepatic de novo lipogenesis by insulin.
SREBP-1c is one of three SREBP isoforms (SREBP-1a, SREBP-1c, and SREBP-2) that belong to the basic helix-loop-helix-leucine zipper family of transcription factors. SREBP-1a and 1c are derived from a single gene using different promoters and exon 1s. The longer acidic NH2-terminus of SREBP-1a makes it more potent than SREBP-1c and can activate all SREBP-responsive genes that mediate the synthesis of fatty acids, TG, and cholesterol. The roles of SREBP-1c and SREPB-2 are more restricted. SREBP-1c preferentially enhances transcription of genes required for fatty acid synthesis, while SREBP-2 preferentially activates genes required for cholesterol synthesis. SREBP-1c and SREBP-2 are predominant isoforms in the liver and most other intact tissues, and SREBP-1c is regulated by insulin [8].
SREBPs are synthesized as inactive precursors that are bound to the endoplasmic reticulum (ER) membrane through two hydrophobic transmembrane-spanning segments connected by a short loop. The NH2-terminal domain of SREBP is the transcription factor region that must be released proteolytically by the two proteases on Golgi apparatus. SREBP cleavage activating protein (SCAP) is required in this process by escorting SREBP from the ER to the Golgi as well as by sensing sterol level in the cell. Other players in the SREBP process are INSIGs (insulin induced genes), which are associated with SCAP. Dissociation of INSIGs from the SCAP-SREBP complex is required for the SREBP-SCAP complex to move to the Golgi [1112]. The released NH2-terminal domain enters the nucleus where it activates multiple target genes by binding to the SREs (sterol response elements) in their promoter (Fig. 2). Enzymes that catalyze the synthesis of fatty acids, TG, and NADPH required for fatty acid synthesis are regulated by SREBP-1c. The typical genes regulated by SREBP-1c are ATP-citrate lyase, acetyl-coenzyme A (CoA) carboxylase, fatty acid synthase, ELOVL6 (elongation of long chain fatty acids family member 6), stearoyl-CoA desaturase, glycerol-3-phosphate acyl transferase, malic enzyme, and glucose 6-phosphate dehydrogenase [8]. The in vivo role of SREBP-1c was demonstrated in a transgenic mouse model that overexpresses SREBP-1c in the liver, which leads to the development of hepatic steatosis due to the increase in lipogenesis [13].
Increased rates of hepatic fatty acid synthesis contribute to the development of hepatic steatosis in rodent models of insulin-resistance and obesity [7]. The leptin-deficient ob/ob mouse is a model of insulin resistance and severe obesity that is commonly used in metabolic studies. Hyperglycemia, hyperinsulinemia, and hyperphagia along with insulin resistance and severe obesity are the representative characteristics of this model. Hyperinsulinemia in ob/ob mice leads to hepatic SREBP-1c activation and hepatic steatosis, while hepatic glucose production is increased due to insulin resistance. The deletion of the SREBP-1c gene in the liver of ob/ob mice results in an approximately 50% reduction of hepatic TG, which indicates a significant role of SREBP-1c in the hepatic steatosis exhibited in the ob/ob mouse, a model of insulin resistance [14]. Compensatory activation of SREBP-2 in the absence of SREBP-1c and the activation of ChREBP by high glucose level are expected for the residual elevation of TG in the liver of this model [15]. ChREBP is a transcription factor that is independently activated by glucose rather than insulin. It activates liver pyruvate kinase, which generates pyruvate, a source of acetyl-CoA, from phosphoenolpyruvate, as well as genes involved in fatty acid synthesis [16].
TARGETING SCAP IN ANIMAL MODELS OF INSULIN RESISTANCE
SCAP is required for the activation of all three isoforms of SREBP. Disruption of Scap in the liver precludes proteolytic cleavage of the NH2-terminal region of SREBPs and abolishes the nuclear forms of all SREBP-1 and -2 in the liver. When Scap is deleted in the liver of normal mouse lines (L-Scap-/-), the expression of target genes in both the cholesterol and the fatty acid synthetic pathways are reduced, and the rates of fatty acid and cholesterol synthesis are diminished by 70% to 80% in the liver [17]. In ob/ob mice, the mRNAs of the genes involved in fatty acid synthesis are markedly elevated because of the SREBP-1c activation. The Scap deletion in the liver of ob/ob mice (L-Scap-/-; ob/ob) abolishes this elevation of lipogenic gene expression, which results in a 90% reduction in fatty acid synthesis rate. The large pale livers in ob/ob mice caused by the massive engorgement with TG are restored to a normal shape. In fact, the hepatic TG contents are completely resolved to normal or even lower than normal levels. The same effects are observed in the insulin resistance model induced by long-term feeding of a high-fat diet. Despite the abolishment of hepatic steatosis in L-Scap-/-; ob/ob mice, the plasma insulin and plasma glucose levels remain high. They fail to improve intolerance to glucose or insulin challenges shown in ob/ob mice with hepatic steatosis. L-Scap-/-; ob/ob mice are one example showing that hepatic fatty acid overproduction is not required for the development of systemic insulin resistance in mice, and hepatic steatosis and insulin resistance may be independent conditions [14].
In humans, dyslipidemia is commonly associated with hepatic steatosis and insulin resistance. Evidence has shown that NAFLD is an independent risk factor of atherogenic dyslipidemia in various clinical studies [1819]. Hamsters easily develop insulin resistance and dyslipidemia after several weeks of being fed a high-fructose diet. As compared with mice, the hamster lipoprotein metabolism more closely resembles that of humans, and high-fructose diets raise plasma very low density lipoprotein (VLDL)-TG and cholesterol levels as well as hepatic TG content. A high-sucrose diet induces hepatic SREBP-1 and the mRNA levels of the genes required for fatty acid synthesis such as acetyl-CoA carboxylase and fatty acid synthase. Depletion of SCAP mRNA by RNAi in hamsters fed a high-sucrose diet for several weeks diminishes SCAP protein in the liver and nuclear SREBPs, which results in the normalization of mRNA levels of those genes. The inhibition of SREBP-1c activation by SCAP RNAi can normalize hepatic TG content as well as plasma VLDL-TG and VLDL-cholesterol in hamsters. The secretion rates of VLDL-TG are reduced by 40%; however, SCAP-RNAi does not significantly affect body weight, plasma insulin level, or blood glucose level in hamsters [14].
CONCLUSIONS
In an insulin-resistant state, the increased influx of fatty acids from adipocytes, the increase in de novo lipogenesis, and reduced fatty acid oxidation result in TG accumulation in the liver. Activation of SREBP-1c, the main transcription factor that increases hepatic fatty acid and TG synthesis, is presented in insulin resistance. Inhibition of SCAP prevents the activation of SREBPs and the expression of genes required for fatty acid and TG synthesis, which results in the inhibition of de novo fatty acid synthesis. As a consequence, the inhibition of SCAP can resolve hepatic steatosis and hypertriglyceridemia in animal models of insulin resistance. Further human studies will reveal whether inhibition of SCAP has the same therapeutic effects in humans as it does in animal models.

 XML Download
XML Download