

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Familial hypercholesterolemia (FH) is a common autosomal dominant genetic disorder and is an important health issue [12]. Patients with FH are exposed to a lifetime burden of high low density lipoprotein cholesterol (LDL-C) and have significantly increased cardiovascular risk. Therefore, early and appropriate diagnosis in affected persons and prevention of vascular complications are critical. In this review, recent studies on the prevalence, diagnosis, vascular risk, and treatment of patient with FH are discussed. Particularly, the focus is on studies of heterozygous FH.
PREVALENCE OF FAMILIAL HYPERCHOLESTEROLEMIA
The frequency of heterozygous FH has long been reported to be 1/500 (0.2%), while that of homozygous FH has been reported to be 1/1,000,000 in many studies conducted in various countries around the world [3]. However, although heterozygous FH is influenced by diagnostic criteria including mutation positivity, the prevalence has been reported to be 1/217 in a very recent Danish study [4] and 1/250 in an American study using Dutch criteria [5].
CLINICAL AND GENETIC DIAGNOSIS
Clinical criteria most commonly used for diagnosis of FH include the Simon Broome Register Group criteria [6], Dutch Lipid Clinic Network criteria [6], and Make Early Diagnosis to Prevent Early Deaths (MEDPED) criteria [7]. A Japanese group has also developed their own criteria for FH [8]. Major items for these clinical diagnostic criteria are (1) severe hypercholesterolemia; (2) physical findings such as xanthoma in the index case; and (3) family history of premature coronary artery disease (CAD) or severe hypercholesterolemia. While the above-mentioned criteria designed in different countries show similarity, they also show difference with respect to specific values (Table 1). For cost-effective and efficient diagnosis, many guidelines and expert groups recommend cascade genetic screening for family members. However, in real practice, diagnosis using only clinical findings from history taking, physical examination, and lipid profile is very common, especially when genetic screening is not available [1].
Most monogenic causes of FH are mutations of low density lipoprotein receptor (LDLR), apolipoprotein B (APOB), or proprotein convertase subtilisin/kexin type 9 (PCSK9) genes. All of these genes are involved in the pathway that clears circulating low-density lipoprotein [9]. Among monogenic FHs, about 90% are caused by mutations of LDLR, for which ≥1,200 different mutations have been identified [1]. However, even family members who have the same mutation often have quite different LDL-C levels, responsiveness to lipid lowering therapy, and survival. Therefore, decision of whether to treat or the intensity of treatment depends mostly on the phenotype rather than genotype. In other words, the clinical importance of genetic diagnosis is limited, and clinical diagnosis is emphasized in real clinical practice [10]. Pathogenic mutations are not identified in about 60% of clinically diagnosed FH patients, although the proportion differs according to the ethnicity or diagnostic criteria. Most FH patients without a monogenic cause are suspected to have polygenic FH that is caused by a multiple lipid-related common variants [2].
Although LDL-C >500 mg/dL or a xanthoma in a child is suspicious for homozygous FH, some patients with homozygous FH have lower LDL-C levels [11].
CARDIOVASCULAR RISK IN FAMILIAL HYPERCHOLESTEROLEMIA
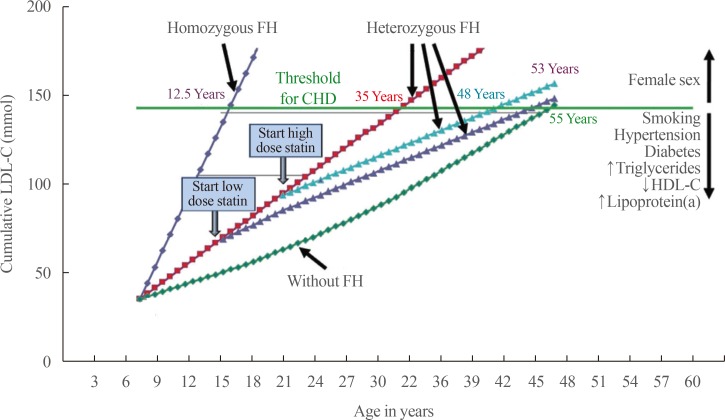
The most critical complication in FH is life-time cardiovascular disease due to longstanding LDL-C burden (Fig. 1). It has been reported that 25% of affected women and 50% of affected men experience cardiovascular complications [12]. The risk of CAD is known to be 3.5- to 16-fold higher in heterozygous FH patients compared to non-FH individuals [13]. The results of studies on the risk of cerebrovascular disease have been inconsistent and the risk is not yet clear [14]. The guidelines recommend that risk assessment in patients with FH should not be made according to European Systematic Coronary Risk Estimation or American Framingham Risk Score, because these scoring systems underestimate cardiovascular risk in FH [1]. Even with the same LDL-C levels, carriers of mutations for FH show higher risk for CAD [15]. In addition, cardiovascular risk in FH is also influenced by other traditional risk factors, and these often also need to be considered when determining treatment [1416].
THE GOALS AND RESULTS OF LIPID-LOWERING TREATMENT AND NOVEL THERAPEUTICS
The main purpose of preventive measures in FH is the reduction of cardiovascular risk. Thus, it is essential to implement pharmaco- and non-pharmacotherapy targeting hypercholesterolemia and other combined risk factors as well. As mentioned above, because vascular risk is very high and associated with cholesterol burden, lowering LDL-C is the mainstay of cardiovascular prevention in FH. Most recent guidelines indicate that it is desirable to reduce LDL-C to 50% of baseline levels or <100 mg/dL in adults with FH. Statins are the first-line agent and ezetimibe or cholesterol-binding resins are recommended when needed [17]. Data from the United Kingdom have demonstrated that the mortality of patients with FH decreased by 25% to 48% after introduction of statins [18]. Earlier treatment is important for efficient prevention of vascular complications. However, because the evidence is limited, there is no sufficient consensus on when to start treatment in children with FH [192021].
Lipoprotein apheresis can be considered in patients with persistently high LDL-C after treatment, extremely high cardiovascular risk, statin intolerance, or especially in patients who have homozygous FH [1]. Meanwhile, the achievement rate of LDL-C goal (<100 mg/dL) is reported to be 11% [14] and a large part of patients do not reach the target. Therefore, PCSK9 inhibitors, recently approved lipid-lowering agents, are spotlighted and expected to meet the unmet medical need (Table 2) [2223]. Although two PCSK9 inhibitors, evolocumab and alirocumab, have shown consistent and strong lipid-lowering efficacy, limitation is their high cost [24]. Lomitapide inhibits microsomal triglyceride transfer protein and reduces very low density lipoprotein (VLDL) assembly, whereas mipomersen blocks the translation of apoB and decreases VLDL synthesis. These two agents were approved for the treatment of homozygous FH. However, drug-related adverse events are relatively frequent and risk/benefit ratio needs to be taken into account for these drugs (Table 2) [2223].
FAMILIAL HYPERCHOLESTEROLEMIA IN ASIA AND KOREA
Although Asian data on FH are not sufficient, its reported prevalence is similar to that of Western countries. The average cholesterol level in Asian patients with FH is likely lower [25]. A recent Korean study suggested that the best cutoff values for predicting mutation-positive FH are total cholesterol 310 mg/dL and LDL-C 225 mg/dL [26]. In Asian patients, 80% to 100% of causing mutations were identified in LDLR, whereas the others were found in APOB or PCSK9 [2526]. Three of 10 Korean FH patients revealed history of CAD. However, the achievement rate of LDL-C by conventional lipid-lowering therapy was not satisfactory [27].
CONCLUSIONS
FH is a relatively frequent genetic disorder. Diagnosis of FH by clinical rather than genetic criteria is more common in real world practice. Reduction of premature complications is most critical in patients with FH. Although, the majority of patients cannot reach a LDL-C goal by conventional pharmacotherapy, novel therapeutics such as PCSK9 inhibitors are expected to improve treatment results.


 XML Download
XML Download