

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Cardiomyopathies, or diseases of the heart that impair its function, can be familial or acquired; their presentation can be classified as arrhythmogenic, dilated, hypertrophic, ischemic, or restrictive. The specific diagnostic criteria for these different forms of heart failure are complex, and the pathogenesis and treatment strategies both vary considerably [1]. These various forms are categorized first by changes in the morphology and function of the heart, often assessed using non-invasive measures such as transthoracic echocardiography, magnetic resonance imaging, or the emerging area of speckle tracking echocardiography. Additional minimally invasive measures may also be used, such as Swan-Ganz catheterization, arterial line catheterization, or transesophageal echocardiography. Each of these approaches provides a robust assessment of cardiac function.
Diabetes mellitus increases the risk of cardiovascular complications. In contrast to other forms of cardiomyopathy, a diagnosis of diabetic cardiomyopathy (DCM) is typically one of exclusion; rather than a specific test, physicians rule out other potential causes of the symptoms. Initially the focus was on the findings of diabetic patients who had heart failure without coronary heart disease. Specifically, DCM is defined as a combination of systolic and diastolic left ventricular dysfunction, pathological left ventricular hypertrophy, and increased interstitial fibrosis [2].
The current review aims to address the factors contributing to DCM by presenting recent advances in determining the causes and mechanisms that play a role in the development of DCM.
DIABETIC CARDIOMYOPATHY: DIAGNOSIS AND PROGNOSIS
It is important to note that controversy remains about the diagnostic accuracy of DCM and whether it exists independently of the type of diabetes [34]. This debate about whether DCM is a valid diagnosis is in part due to the inconsistencies in the definition. A potential argument against the current criteria is that subclinical myocardial dysfunction that is not progressive over time and does not result in chamber enlargement, hypertrophy, or clinical heart failure is not necessarily indicative of a cardiomyopathy [3]. Despite this, the hypothesis persists that diabetes, and particularly fluctuations in glycemic control, alter disease susceptibility. In the case of type 1 diabetes, continuing follow-up of the patient cohort from the Diabetes Control and Complication Trial by the Epidemiology of Diabetes Intervention and Complications group has found that even 30 years later, in those who had undergone intensive therapy to control blood glucose levels there was a decrease of approximately 30% in the incidence of cardiovascular disease and major events including myocardial infarction or cardiovascular death [5]. Similarly, a long-term follow-up study of type 2 diabetes patients assigned to tight glycemic control revealed fewer major cardiovascular events [6]. These studies support the unified hypothesis that prior changes in glucose levels and delivery may lead to persistent molecular changes predisposing patients to the development of DCM.
Owing to the importance of regulating glucose levels as highlighted above, many studies have focused on the signaling pathways that control glucose uptake, storage, and utilization. The most prominent area of research is probably on insulin resistance, which plays a major role in the development of cardiac dysfunction by promoting cellular dysfunction. This includes uncoupling of mitochondria, disrupted autophagy, increased inflammation, impaired Ca2+ handling, and apoptosis. The resistance of cardiac cells to properly respond to a cascade of molecular changes involves well-established factors such as the transcription factor Foxo1 (Forkhead box protein O1), which in turn has a pivotal role in the regulation of genes for metabolism, oxidative stress, inflammation, and apoptosis, as recently reviewed in detail [7]. In healthy individuals, postprandial increases in insulin stimulate the uptake of glucose from the bloodstream and initiate anabolic metabolism and energy storage. In contrast, following a fast, other hormones such as glucagon signal for catabolic metabolism to generate adenosine triphosphate (ATP) [8]. These pathways do not work in isolation, and the crosstalk between the insulin receptor and the β-adrenergic receptor has promoted speculation about combined approaches to protect cardiac contractile function in DCM. This specific topic has very recently been reviewed in detail elsewhere [9].
CAUSES OF DIABETIC CARDIOMYOPATHY
Diabetes mellitus is characterized by high levels of circulating glucose during fasting, although the cause of this differs between the types of diabetes. Type 1 diabetes, also termed insulin-dependent diabetes mellitus, refers to the autoimmune destruction of β-cells in the pancreas, which leads to insufficient production of insulin. Type 2 diabetes, also referred to as non-insulin-dependent diabetes mellitus, is defined by a loss of insulin sensitivity, and is associated with poor diet, obesity, and inactivity. Diabetic patients often present with comorbidities such as hypertension and coronary artery disease. A question that has persisted is the specific mechanism(s) of how diabetes contributes to myocardial dysfunction in particular. It is well-documented that patients with type 2 diabetes are at an increased risk for heart failure. Diabetic patients develop heart failure more frequently and at an earlier age [10]. Because patients often present with multiple risk factors, it is difficult to define the extent to which diabetes contributes to heart failure disease progression. Nonetheless, there are both modifiable and non-modifiable risk factors that will be discussed below.
Modifiable risk factors
Diet and exercise are modifiable factors that are extremely important for cardiovascular health [11]. Even with an appropriate diet, a sedentary lifestyle in itself can have a detrimental effect on glucose uptake and heart health. Both diet and exercise are important in maintaining general health, and poor daily decisions can lead to chronic diseases such as DCM. Diet and exercise are perhaps the easiest to modify among the modifiable risk factors for diabetes and heart failure. However, social interactions can make a proper diet and adequate exercise difficult to achieve. In particular, socioeconomic status plays a large role in diet, exercise, and heart healthiness in general [12]. Though policy reforms and education can modify socioeconomic status, this risk factor can be difficult to directly alleviate, particularly in large populations. Stress is another modifiable risk factor for diabetes and heart failure that is not easy to treat quickly [13]. In this chronic disease, complete lifestyle changes are necessary to drastically improve a patient's prognosis. Nonetheless, in a chronic state of disease, even small changes can be very meaningful for extending and improving the quality of life. For example, simply breaking up the length of sedentary time periods can improve circulating metabolites [14], and a number of additional studies have indicated that reduced cardiometabolic risk and improved glycemic control can result of changes as simple as movement breaks to break up sedentary lifestyles [15].
Non-modifiable risk factors
The modifiable risk factors described above can be exacerbated by non-modifiable risk factors such as age, race, and sex. Some populations with a genetic susceptibility to developing diabetes and heart failure include Asians and African Americans. For example, a specific genetic variant in the aldehyde dehydrogenase 2 family (mitochondrial; ALDH2) was significantly associated with metabolic syndrome, meaning that affected individuals are at a higher risk of cardiovascular disease and diabetes [16]. Furthermore, the Korean Genome and Epidemiology Study recently identified two single nucleotide polymorphisms (SNPs) in glucokinase (GCK) and YKT6 v-SNARE homolog (YKT6) in the Ansung and Ansan cohorts that were significantly linked to prediabetes [17]. A recent review has highlighted some of the top genetic variants that may help explain the high prevalence of type 2 diabetes in African Americans [18]. There are numerous more examples in other racial subpopulations, but together they all highlight the importance for healthcare providers of realizing that patients require precision therapies and ensuring that modifiable risk factors are addressed to the maximal extent while working to minimize the effects of non-modifiable risk factors. Furthermore, epigenetic changes can even serve as an indicator of patients who are progressing to a diseased state, as we have recently reviewed [19]. Briefly, epigenetics includes the modification of gene expression without altering the genetic code. These modifications include DNA methylation, histone modifications (e.g., acetylation), and non-coding RNAs. They are in stark contrast to the SNPs described above and represent the interplay between environmental influences and the genome. A complete understanding of the epigenetic changes that can occur throughout life needs to be achieved to personalize medicine and to accurately establish the cause of DCM. One area that has received particular focus owing to its promise for therapeutic intervention is that of microRNAs, reviewed elsewhere [20]. It is evident that the true cause of the chronic disease state of DCM will vary across all patients. However, with more knowledge of general trends, epigenetic changes may function as modifiable risk factors.
Epidemiological studies have shown that diabetes is correlated with the potential for accelerated disease progression in certain populations pertaining to glycemic control [5]. The consensus holds that non-diabetic patients can have severely decreased cardiac function, but that it is reasonable to conclude that diabetes is maladaptive in heart failure. Treating diabetic patients with heart failure is an individually unique challenge. There are overlapping risk factors for diabetes and heart failure, which mostly have to do with substrate availability. The lack of oxygen in ischemia and the excess nutrients in diabetes contribute to the milieu that makes DCM so deadly.
MECHANISMS OF DIABETIC CARDIOMYOPATHY
In the prior section, we identified broad concepts that have been attributed to DCM. In this section, we will focus on specific molecular mechanisms contributing to disease progression. Noticeable changes occur at a systemic level, such as decreased cardiac contractility, hyperglycemia, and increased cardiac inflammation. Additional changes are present at the cellular level, such as increased fibrosis and fatty acid utilization. Finally, there are changes at the molecular level, such as alterations in gene expression, signaling, and protein levels. The switch between extremes at each level—organismal, cellular, and molecular—combine to add increased stress on cardiac tissue, which leads to DCM.
At the organismal level, increased cardiac rigidity alters cardiac contractile function, blood pressure, and output. There are a number of cellular and molecular mechanisms that have been proposed to contribute to this decline in function, including endoplasmic reticulum stress, impaired autophagy, increased apoptosis/necrosis, inflammation, and altered insulin signaling [21]. Due to a decrease in insulin sensitivity, there is less glucose uptake, and this in turn contributes to the development of hyperglycemia. This in turn can exert feedback on molecular regulation through the generation of advanced glycation end products (AGEs). These glycosylated proteins crosslink collagen molecules, which increases fibrosis because the collagen cannot be degraded [21].
DCM is also associated with an increase in reactive oxygen species (ROS), leading to DNA damage and even an increase in AGE receptors. The hyperlipidemia seen in diabetic patients has been shown to increase stress on the endoplasmic reticulum due to increased protein production and to increase stress on the mitochondria as they attempt to generate enough ATP. This increase in production leads to increased ROS levels [22]. There have been several studies showcasing the benefits of using antioxidant drugs in patients with cardiovascular injuries [232425]. The carotenoid bixin possesses anti-inflammatory and antioxidant activities, and was shown to inhibit cardiac fibrosis and to decrease proinflammatory cytokines. It also decreased oxidative stress triggered by high fat by activating the nuclear factor-E2-related factor 2 (Nrf2) pathways [23]. Mito-TEMPO ((2-(2,2,6,6-Tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl)triphenylphosphonium chloride) was shown to reduce ROS, decrease apoptosis, and decrease hypertrophy in both type 1 and type 2 diabetes [24]. The flavonoid quercetin-3-O-rutinoside (Rutin), ameliorates the reduced antioxidant capacity, increased inflammation, and degenerative changes in mice with streptozotocin-induced diabetes [25]. Nrf2, Mito-TEMPO, and Rutin all show promise in reducing factors that contribute to the formation of DCM.
There is a tight connection between DCM and metabolic dysfunction in the mitochondria, which causes the shifting of carbon placement and through uncoupled respiration increases the presence of ROS. Most of the ROS in a failing heart come from the mitochondrial electron transport chain, and ROS signaling has been shown to be involved in maladaptive hypertrophy, contractile dysfunction, and damage to mitochondrial DNA. This mitochondrial DNA damage affects energy output and can lead to cardiomyocyte death [26]. The cycle of mitochondrial dysfunction and ROS production can lead to larger-scale issues such as cell death and fibrosis, which are major contributors to DCM. It has been shown that an increase in signaling pathways, such as transforming growth factor-β, can increase apoptosis in cardiomyocytes [21]. Another signaling molecule, vascular endothelial growth factor B (VEGFB), is located on the surface of cardiomyocytes and is released to signal survival gene expression in response to high glucose. However, it has been shown that in diabetic patients, cardiomyocytes are unable to respond to VEGFB, leading to an increase in apoptosis [27].
CELLULAR UPTAKE AND UTILIZATION OF MOLECULAR FUELS
The heart is a metabolic omnivore that can utilize available substrates for energy production. In utero, the heart primarily consumes glucose as its main source of energy. After birth, the heart switches to primarily utilizing fatty acids [28]. In a typical failing or aging heart, there is a reversion back to the utilization of glucose, but a diabetic heart does not make this switch. It is believed that the heart's inability to maintain the flexibility to use both glucose and fatty acids leads to cardiac failure.
Glucose is transported across the cell membrane via glucose transporters (GLUTs); including both insulin-regulated and constitutive uptake by the major cardiac variants GLUT4 and GLUT1, respectively. In cardiac tissue the expression of GLUT4 is four times higher than that of GLUT1 [29]. Cardiac tissue primarily utilizes GLUT4 for its glucose uptake, but in diabetic patients there is a decrease in expression of GLUT4 and therefore a decrease in the uptake and utilization of glucose. When GLUT4 is overexpressed systemically, there is a rescue of cardiac contractility and metabolism [30]. However, when increased GLUT1 expression is induced in the presence of diet-induced obesity, cardiac dysfunction is enhanced [31]. Therefore, caution must be taken when considering altering glucose delivery as a therapy, and additional work should also explore the potential of increasing glucose delivery in the context of pre-existing diabetes.
Fatty acids are hydrophobic and therefore not water-soluble. Therefore, when entering the heart the fatty acids are either attached to albumin or as lipoproteins [32]. In diabetic patients, there is an increase in fatty acid oxidation (FAO) in comparison to carbohydrate oxidation [33]. There is an increase in FAO from 70% in normal cardiomyocytes to 80% to 90% in the diabetic heart [34]. One potential therapy that has received considerable focus has been the use of metformin. Although some early concerns were raised regarding the effects of long-term use on the heart, in a systematic review of 17 studies, metformin improved clinical outcomes even in patients with moderate congestive heart failure [35]. Additional new therapies that alter metabolism include antioxidants such as aspalathin, a dietary flavonoid that was found to work as well as metformin in improving fatty acid uptake in a cell culture model of glucotoxicity [36]. This compound and other drugs have the potential for use as adjuvant therapies to provide increased cardiac benefits when treating diabetes.
In the liver, ketone bodies are synthesized using acetyl-CoA, which is generated from FAO. In a fasting state, the concentration of ketone bodies in circulation increases, and this increase in concentration is even greater in diabetic ketoacidosis. The metabolism of ketone bodies helps maintain bioenergetic homeostasis when there is limited bioavailability of carbohydrates. They also help regulate metabolism and ROS production, which could indicate a role in signaling in cardiomyocytes [37].
Previous studies have shown that reprogramming of gene expression occurs, which can contribute to altered reliance on specific metabolic substrates [19]. The heart may adapt to some of these changes in order to maintain the energy required to carry out necessary functions, such as continued blood flow. One hypothesis is that the heart becomes increasingly more reliant upon anaplerotic pathways to supply substrates to replenish intermediates of the citric acid cycle [38]. The primary anaplerotic reaction is the generation of oxaloacetate from pyruvate by pyruvate carboxylase [37]. While pyruvate carboxylase has not been extensively studied in cardiac tissue, it plays the important role of converting pyruvate into oxaloacetate, which allows the cell to bypass the conversion of pyruvate into acetyl-CoA by pyruvate dehydrogenase (PDH). This ability to compensate is important, as PDH activity is decreased by elevations in pyruvate dehydrogenase kinase 4 (PDK4), which has been shown to be increased in the heart by diabetes in an insulin-dependent manner [39]. The role of PDK4 in disease progression has led researchers to target its mechanism of action in obesity, diabetes, heart failure, and vascular calcification [40].
IMPACT OF HORMONES AND SIGNALING
Several hormonal changes occur in diabetic patients, particularly the trademark hormone of the disease: insulin. Insulin levels are known to be high in type 2 diabetic patients and absent in type 1 diabetic patients. These trends can vary depending on the stage of the disease, with particularly higher levels in the early stages of type 2 diabetes as the pancreas tries to compensate for hyperglycemia, although glucose uptake is decreased due to a loss of sensitivity. Glucose uptake is diminished in patients exhibiting decreased glucose tolerance in segments of the heart with preserved perfusion. In contrast, glucose uptake was found to be increased in segments of the heart with decreased perfusion in prediabetic patients, most likely because the physiological response of hypoxia induced glucose uptake [41]. Insulin sensitivity is lost in the hearts of diabetic patients, but it is known that glucose is still utilized, as GLUT4 is stimulated by insulin and muscle contraction [42]. Despite decreased glucose uptake in the diabetic heart, there is still an excess in circulating glucose levels, and how circulating glucose levels impact the heart is an area of active research. It is possible that glucose-mediated damage contributes to the decline of cardiac function in a process termed glucotoxicity. This is supported by the finding that increasing urinary glucose excretion with sodium glucose cotransporter-2 inhibition provides cardiac protection [43]. It can be ascertained that glucose continues to play a significant role in the diabetic heart, but perhaps through non-oxidative pathways, as discussed elsewhere in this paper (e.g., O-linked β-N-acetylglucosamine [O-GlcNAc] addition and AGE).
Heart failure in itself can lead to hormonal changes in the renin-angiotensin-aldosterone system (RAAS); these complexities will be explored here, as they directly relate to DCM disease progression. RAAS can cause systemic blood pressure increases and intracellular changes in cardiomyocytes that contribute to oxidative stress [44]. It was shown in diabetic rat hearts that angiotensin 2 receptors were increased in cardiomyocytes, which led to a physiological decrease in left ventricular systolic pressure but an increase in diastolic pressure [45]. This indicates that the molecular changes that occur in cardiomyocytes due to hyperglycemia alter the heart's ability to respond to stimuli. This particular hormonal change suggests that diabetic hearts lose structural and molecular flexibility. Angiotensin receptors in the heart are known to cause cardiac restructuring; thereby, allowing for the subsequent increase in blood pressure [46]. Furthermore, hyperglycemia alone can increase blood viscosity via an increase in capillary permeability and a concomitant increase in hematocrit [47]. This increase in osmolarity has been associated with a decrease in cardioprotection with ischemia reperfusion injuries [48]. Oxygen and blood supply have been discussed here as factors contributing to cardiomyopathy. In diabetic patients, glucose begins its damage to the cardiovascular system in the blood, and the hormonal changes that result contribute to cardiomyopathy development.
Inhibitors of angiotensin-converting enzyme (ACE), a central component of RAAS that controls blood pressure, have been proven to be an effective treatment in congestive heart failure patients. For example, a 2005 study in streptozotocin-treated rats revealed that ACE inhibitors reduced superoxide levels in the diabetic myocardium [49]. This study also found that oxidative stress was induced, at least in part, by RAAS activation. Diabetes-induced RAAS activation contributes to a decrease in cardiac function through inflammation, fibrosis, and oxidative stress. These factors, which are caused by hormonal changes, inhibit the heart's ability to function. In diabetic patients, hormonal therapeutics may have a future as treatments of cardiomyopathy. The hormonal changes occur in response to hyperglycemia. If it can be ascertained that glucose stimulates RAAS, the response of this system in diabetic patients is a normal physiological process that is ultimately maladaptive in that it contributes to the development of DCM.
OTHER FACTORS CONTRIBUTING TO DIABETIC CARDIOMYOPATHY
The most significant comorbidities of diabetes arise from the trademark sign of diabetes: hyperglycemia. Chronically high levels of circulating glucose have extreme implications on the body as a whole, primarily contributing to microvascular dysfunction, which manifests as retinopathy, neuropathy, and nephropathy. For example, the metabolic milieu that arises from diabetes can contribute to atherosclerosis because of the high circulating levels of glucose, lipids, and cholesterol. High circulating levels of these metabolites can stick to the walls of arteries, and this also plays a role in the development of cardiomyopathy via ischemia. Furthermore, hyperglycemia and hyperlipidemia activate the immune system, which mediates more damage to the lumen of arteries [50]. This vascular damage can cause a decrease in oxygen delivery in the heart. This damage contributes to the role of diabetes as a risk factor for ischemic heart disease [51]. Even in undiagnosed patients, hypoxia is a problem that diabetic patients must deal with because of damage to the vasculature [41].
In prediabetes, myocardial metabolic compensation may occur to compensate for the hyperglycemia that presents in diabetic patients. Lipotoxicity can occur when the high levels of fatty acids are not all oxidized for fuel, and instead enter non-oxidative pathways [52]. Insulin plays a role in mediating glucose and fatty acid uptake. Clinically, glucose uptake is known to be decreased in the diabetic heart [41]. It is hypothesized that lipid uptake is exacerbated because of the oversupply and that lipids therefore accumulate in the cell, leading to further complications in the diabetic heart [53]. The healthy heart does prefer fatty acids, but the metabolic imbalance in diabetes causes the heart to lose its flexibility in utilizing substrates. This loss in flexibility is believed to lead to poor heart outcomes in diabetic patients. Aside from glucose and fatty acids, ketone bodies are a substrate that the failing heart appears to preserve the capacity to oxidize [5455].
Glucose uptake and oxidation are tightly linked with oxygen availability. In insulin-resistant patients, glucose uptake is increased in segments of the heart that have decreased blood flow [41]. The decreased blood flow is likely due to atherosclerotic plaques in coronary arteries. Hypoxia stimulates glucose uptake, but also shunts glucose into glycogenesis [56]. Insulin-resistant patients also showed a decrease in glucose uptake in segments of the heart with preserved blood flow [41]. It is known that a decrease in glucose uptake can increase the amount of glucose that enters the non-oxidative hexosamine biosynthesis pathway (HBP) [57], but this mechanism is mediated by intracellular calcium levels. Perhaps contraction itself is a regulator of the end product of HBP, O-GlcNAc, which modifies proteins in a similar fashion as phosphorylation. This can lead to an increase in O-GlcNAc, which can be adaptive in an acute postischemic state [58]. The problem is that chronic O-GlcNAc increases can be maladaptive, as seen in pressure overload heart failure and DCM [59]. Oxygen availability and glucose utilization can become imbalanced in diabetic patients. In diabetic patients, a decrease in glucose uptake with preserved oxygen supply should lead to more glucose being utilized for O-GlcNAc modifications. An increase in glucose uptake with diminished oxygen supply should also lead to a change in O-GlcNAc levels. The oxygen supply here may be linked with O-GlcNAc modification, so as to mediate signaling and either to prevent or to promote ROS generation for cell signaling. In the diabetic heart, it may be the case that oxygen availability plays a crucial role in the metabolic changes that are seen in patients with DCM.
CONCLUSIONS
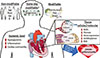
The connection of diabetes to cardiovascular disease has been known for decades. Nonetheless, controversy persists in defining the disease as a single entity, and the mechanisms of how diabetes alters cardiac function are incompletely understood. Fig. 1 contains a summary of what has been consistently established concerning known modifiable and non-modifiable risk factors that should be focused on when trying to prevent and reverse the development of heart failure in this susceptible population. Exciting new genome-wide association studies are continuing to identify genetic susceptibilities that may help to explain the diverse susceptibility to heart failure in diabetic patients and to provide novel treatments for non-modifiable risk factors. The rapidly emerging area of epigenetics provides a novel area of potential future interventions, as we define the signaling pathways regulating these changes and better characterize the mechanisms by which these marks are placed, maintained, and potentially reversed. The successful pursuit of combining these new areas with the vast knowledge base already available may still provide hope for tackling the devastating complications associated with diabetes, both in the heart, as described here, and potentially in other organs as well.
 XML Download
XML Download