

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Metabolites are typically low molecular weight (<1,500 Da) biomolecules. They are the building blocks of the genome, proteome, and cell membranes and play key roles in biology as signaling molecules, energy sources, and metabolic intermediates. Their levels provide integrative information on biological functions and define the phenotypes of biological systems in response to genetic or environmental changes (Fig. 1). Metabolite analysis involves the use of different analytical techniques to identify and quantify intracellular and extracellular metabolites. Successful metabolomic studies rely on proper sample preparation, innovative instrumentation, and bioinformatic tools [1]. Metabolites show broad variations in physicochemical properties, such as polarity, acidity, and volatility. Thus, it is difficult to simultaneously analyze a broad range of metabolites [2].
Metabolomics has developed along with innovative analytical instrumentation. Various analytical techniques have all been applied for metabolite analysis, including mass spectrometry (MS), nuclear magnetic resonance (NMR), Fourier transform infrared (FT-IR) spectroscopy, and Raman spectroscopy (Fig. 2) [3456789]. FT-IR spectroscopy and Raman spectroscopy are rapid, nondestructive, and high-throughput analytical methods that be used for various metabolites. These spectroscopic methods are generally applied to profile metabolic alterations due to their holistic nature, and absorptions at specific wavelengths are sometimes able to provide important clues for the identification of observed metabolic components. However, the sensitivity and selectivity of FT-IR spectroscopy and Raman spectroscopy are lower than those of other methods [8]. NMR spectroscopy, which is a commonly used strategy in metabolomics, is also a rapid and nondestructive method with minimal sample preparation. Chemical shifts in NMR spectra can provide crucial evidence for the identification of specific metabolites. However, NMR requires at least millimolar concentrations of metabolites in samples and its sensitivity is lower than that of MS. Another major disadvantage of NMR is its poor dynamic range [7]. On the other hand, MS remains the most favorable technology for metabolomics due to its wide dynamic range and good sensitivity (nM). In addition, MS is able to detect a diverse range of molecular species comparing to other strategies.
MS-BASED METABOLOMICS
Recently, MS-based metabolomics became the most popular metabolomics strategy. To establish metabolomic platforms, separation modules are often connected to the MS system. Liquid chromatography/MS (LC/MS) has several advantages over MS alone. Ion suppression, which is often caused by a complex biological matrix, can be eliminated because a metabolite can elute at different retention times with any disturbed metabolites. In addition, the chromatographic peak area provides a useful tool for metabolite quantification because accurate quantification cannot be obtained when MS is used alone.
Gas chromatography/MS (GC/MS) has been the most suitable MS method for the detection of volatile metabolites. This technique often requires chemical derivatization to improve volatility, and its application has limitation in terms of the molecular size, volatility, and polarity. Therefore, LC/MS has become a popular choice in metabolomics. Sample derivatization is generally not required in LC/MS, and metabolites with more diverse chemical structures and increased molecular sizes can be measured [4810].
MS-based metabolomics may have difficulties in the identification and characterization of observed metabolites. Mass spectral libraries have not been complete enough for identification of all observed metabolites. The Golm metabolome database consists of mostly plant metabolites, and the National Institute of Standards and Technology database includes only electron ionization-MS data [11]. The METLIN database contains an annotated list of metabolite structural information with MS; however, the database is still not sufficient to identify all observed metabolites [12]. Recently, the Human Metabolome Database (HMDB) was also established to provide chemical and molecular biochemistry data on human metabolites [13]. In addition to the previously mentioned web-based databases, metabolite identification has been reported by commercially available or in-house databases. However, the existence of isobaric or isomeric metabolites may result in misidentification. MS/MS involving collision-induced dissociation has been used to obtain structural information on metabolites [514151617]. The characteristic fragmentation pattern of a metabolite is important for the identification of unknown metabolites and analytical specificity. In addition, the specific retention time in certain separation conditions or the isotopic patterns of unknown metabolites should provide clues for metabolite identification and structural information.
NONTARGETED METABOLOMICS
Nontargeted metabolomics aims to profile the entire metabolome present in cells, biofluids, or tissues. Nontargeted metabolomics measures as many metabolites as possible to compare biological samples and is useful for hypothesis generation. However, the greatest bottleneck in nontargeted metabolomics is the identification of unknown features [18]. Metabolites are found in low picomolar to millimolar concentrations with various physicochemical properties. Thus, it is impossible to observe all metabolites present in a biological sample. Sample preparation and proper analytical instrumentation are important to detect the maximum number of metabolites. High-resolution MS techniques such as time of flight, Orbitrap, or Fourier transform ion cyclotron resonance should be used in nontargeted metabolomics because accurate mass determination is crucial in metabolite identification [1].
Global metabolome screening is highly reliant on state-of-the art instruments, bioinformatic tools, and software. General procedures to perform global metabolome profiling involve sample preparation, instrumentation, and statistical or bioinformatic analysis and have been described in several prestigious journals [192021]. General workflow for global metabolome profiling can be found in Fig. 3.
Sample preparation can be performed to reduce the sample matrix effect and improve sensitivity. Metabolites in a biological sample can sometimes be divided into aliquots of similar physicochemical properties. Liquid-liquid extraction is often used to prepare a sample into two groups of either hydrophobic metabolites or hydrophilic metabolites. Sometimes, simple filtration can be performed to remove solid matter from biological samples, particularly urine. Deproteinization with organic solvents is commonly performed for biofluids.
Metabolic profiling studies have been performed using various analytical platforms such as capillary electrophoresis (CE)/MS, GC/MS, and LC/MS. Any single analytical platform cannot observe all metabolites in a sample due to the complexity of the concentrations and physicochemical properties of the metabolites. To increase the coverage of observed metabolites, multiple analytical platforms should be used for global metabolome profiling [2223]. GC/MS is usually the preferred platform for small and volatile metabolites, including steroids and fatty acids, and chemical derivatization is often performed to increase the volatility of metabolites. LC/MS can detect a broader range of metabolites, regardless of their hydrophilic or hydrophobic nature, and CE/MS is more appropriate than other platforms for detecting hydrophilic metabolites [24]. Although multiple analytical platforms are recommended for nontargeted metabolomics, not all laboratories have access to all of these analytical instrumentations. Thus, in many cases, the analytical platforms used are chosen depending on their availability and the application.
Global metabolome profiling data should be acquired from both positive and negative ion mode when the LC/MS or CE/MS platform is used. Positive ion mode can detect basic metabolites, whereas negative ion mode can better detect acidic metabolites. Thus, the maximum coverage of metabolites can be obtained when mass spectrometric data are acquired from both positive and negative ion mode.
Accordingly, the sample condition and preparation, chromatographic separation, and type of mass analyzer should be considered to obtain high-quality data for global metabolome profiling. The data acquired by global metabolome profiling consists of a list of the mass to charge ratios (m/z) of intact metabolites metabolites and their retention times. Metabolomic software performs data processing and provides such features as P values and fold changes between samples. XCMS and MZmine are freely available web-based interfaces for metabolomic data analysis [252627]. To identify the unknown metabolites of interest, the accurate mass of the metabolite is used to find matches in the metabolite database, such as HMDB and METLIN. Database matches may not be found for many of the observed peaks, and sometimes numerous hits for one peak can be found, even with a narrow range of mass accuracy (often 10 ppm). Correct assignment can be done by comparing the retention time and MS/MS data. Because small molecules can share the same mass and elemental composition, database search results cannot guarantee the correct identification of observed metabolites and the results have to be confirmed with other strategies such as targeted metabolomics. The metabolites identified can be subjected to biological pathway analysis to find relationships with specific disorders or physiological characteristics using web-based databases such as Kyoto Encyclopedia of Genes and Genomes (KEGG), MetaCyc, and BioCyc [282930].
TARGETED METABOLOMICS
Targeted metabolomics provides quantitative information for a predefined list of metabolites or metabolic pathways of interest and is useful for answering any specific biochemical questions or hypotheses [313233]. Each targeted platform is composed of the known metabolites, and the specific analytical method for each platform is established using authentic standards or chemically synthesized compounds. The quantitative information provided by targeted approaches is reliable and has better sensitivity than nontargeted approaches. Without specific analytical platforms, it is often difficult to observe low-level metabolites, namely, those that are present at a low concentration in a biological system. Thus, targeted metabolic platforms are especially preferable for low-level metabolites, which are often bioactive and play important roles in biological systems. Also, when the main interest is a specific class of metabolites or a metabolic pathway, targeted approaches can provide better insight into the specific goal. Targeted metabolic analysis can be a benefit for some metabolites that are not observed well due to their molecular nature, especially when easily ionizable metabolites or isobaric compounds are present.
It has been reported that the human serum metabolome contains about 20 chemical classes, although other classifications are also possible depending on one's perspective [23]. Different analytical methods have been established to specifically detect each class of metabolite or metabolites related to a metabolic pathway [234]. Commonly, GC or LC separation conditions and different MS/MS techniques can be used to distinguish isobaric or structurally very similar metabolites [35].
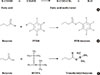
As an example, targeted analytical platforms of short-chain fatty acids, fatty acids, and eicosanoids will be explained in the following paragraph. Short-chain fatty acids (e.g., acetate, butyrate, propionate, and lactate) are those fatty acids with less than six carbons. They have different chemical and biological properties than fatty acids with more than six carbons. Longer fatty acids are less volatile and less water soluble than short-chain fatty acids. Thus, the extraction protocol should depend on the type of fatty acid. Extraction of short-chain fatty acids from biological samples involves the incubation of the samples with water and uses the aqueous phase for analysis [36]. On the other hand, organic phase extraction should be performed to obtain longer-chain fatty acids from biological samples. Methyl esterification using methanol with BCl3 is a commonly accepted chemical derivatization method for longer-chain fatty acids, but short-chain fatty acids have been derivatized by several methods, such as BSTFA (N, O-bis(trimethyl-silyl) trifluoroacetamide) for trimethylsilylation or PFBB (2,3,4,5,6-pentafluorobenzyl bromide) for PFB (pentafluorobenzyl)-linked derivatives (Fig. 4). These fatty acid derivatives are separated using a GC column, with optimized column type and temperature gradient, then ionized using an electron impact source and analyzed with MS. Intact molecular ions are rarely observed due to the characteristics of the electron impaction source, and specific fragment ions for each fatty acid are used for identification and quantification.
Eicosanoids are oxidized 20-carbon fatty acids that play key roles in inflammation and immunity. The levels of eicosanoids and their balances affect various biological processes and diseases. Fatty acids in human serum are present at micromolar concentrations, but most eicosanoids in human serum are found at nanomolar or lower concentrations. Thus, the use of solid phase extraction is well established for capturing eicosanoids from complex biological matrixes. Eicosanoids have similar chemical structures, and some of them are isobaric. Accordingly, LC separation is very important to distinguish different eicosanoids among isobaric compounds [37]. Prostaglandin D2 (PGD2) and prostaglandin E2 (PGE2) are isobaric and their elemental compositions are the same. The only difference between PGD2 and PGE2 is their stereochemistry. Careful optimization of LC conditions permits separation of PGD2 and PGE2, as shown in Fig. 5A. About 40 eicosanoids can be analyzed using the LC-MS/MS system, in which the multiple reaction monitoring mode is applied to specifically detect each eicosanoid (Fig. 5B).
As demonstrated, targeted metabolome platforms should be established to separate target metabolites for better ionization and quantification using sample preparation and a separation module, and to specifically detect metabolites from complex biological matrixes using characteristic fragment ions generated by MS/MS. Greater effort is required in targeted approaches, but targeted metabolome platforms provide absolute or relative quantitative data for specific metabolic pathways once the platform is successfully established.
Targeted metabolic profiling without standard metabolites is also useful to understand biology or disease states. Phospholipid profiling is a good example. The headgroups of phospholipids are easily observed in positive ion mode during collision activated dissociation, which is a commonly used MS/MS technique, whereas fatty acyl chains are observed in negative ion mode. Thus, LC-MS/MS analysis in both positive and negative ion modes provides structural information on phospholipids. Accordingly, a specific MS/MS technique known as neutral loss scan or precursor ion scan is able to profile a certain class of phospholipids, and relative quantification is also possible by comparing the peak areas of masses corresponding to specific phospholipids.
METABOLOMICS APPLICATIONS IN BIOMEDICAL RESEARCH
Metabolites are substrates and products of diverse biochemical reactions and commonly related to the basic biology of the genome and proteome. Metabolomics aims to explore chemical–biological interactions and understand the roles of metabolites in basic, translational, and clinical research. Metabolites have played very important roles in clinical applications, with more than 95% of clinical assays using metabolites [38]. The most representative example would be determination of the blood glucose level for diabetes patients. Most common drugs used today are still small molecules, and more than half of them are derived from metabolites [39]. In addition, human diseases are related to unfavorable interactions of the genome or proteome with metabolites, and a disordered metabolism is involved in many genetic disorders. Recent developments in analytical platforms that use MS have enabled metabolomics to become an important strategy in biomedical research.
One of the earliest studies using metabolomics in diabetes was plasma phospholipid metabolic profiling for class separation between diabetes mellitus type 2 and the control [40]. This study showed that metabolomics could be used in class separation using multivariate statistical analysis such as partial least square discriminant analysis and principal component analysis and provide potential biomarkers for discrimination. Some groups subsequently performed metabolic studies using both the LC-MS/MS system and H-NMR (proton nuclear magnetic resonance spectroscopy) NMR to explore metabolic pathways altered by insulin deficiency. Two analytical platforms revealed that several metabolic pathways, such as amino acid oxidation, mitochondrial bioenergetics, and gluconeogenesis, were perturbed during insulin deficiency [41]. Newgard et al. [42] used metabolomic profiling to reveal that a branched-chain amino acid-related metabolic signature differentiates obese and lean humans, and contributes to insulin resistance. Through metabolite profiles of human subjects, Wang et al. [43] found five amino acids that can predict an earlier risk of diabetes development. They performed metabolite profiling on the samples of control and prediabetic groups matched according to sex, age, body mass index (BMI), and fasting glucose during a 12-year follow-up period. Branched-chain amino acids (leucine, isoleucine, valine) and aromatic amino acids (phenylalanine, tyrosine) were significantly altered in the prediabetic group. They also measured these five metabolites in a randomly chosen control group that was not matched according to sex, age, BMI, and fasting glucose, confirming the significance of these metabolites. In addition, they assessed the predictive performance of the combination of the five amino acids for the risk of diabetes. In their study, they used a targeted metabolomics strategy, measuring amino acids, urea cycle metabolites, and nucleotide metabolites. On the other hand, Zhao et al. [44] used nontargeted metabolomics to investigate the metabolic signatures of prediabetic subjects in both urine and plasma. They found that fatty acid, tryptophan, uric acid, bile acid, and lysophosphatidylcholine metabolism were the prediabetic-associated alterations.
Fibrosis develops differentially in different individuals, and there has been no report of any reliable factors that can predict fibrosis susceptibility. Mitochondrial dysfunction has been suggested to be closely related to fibrosis through metabolic perturbations, with Maeda [45] reporting that a radiation-induced fibrosis model had mitochondrial homeostasis problems. Lipid accumulation, including that of ceramides, diacylglycerols, and sphingomyelins, was caused by reduced fatty acid oxidation. This observation led to the possible use of metabolomics to find predictive or diagnostic markers. The plasma fatty acid composition was different in both cystic fibrosis patients and animal models. Bile acids for lipid absorption and cholesterol metabolism were altered in cystic fibrosis, which indicates the imbalanced lipid metabolism in cystic fibrosis [4647]. In addition, sphingosine-1-phosphate was increased due to upregulation of sphingosine kinase in human fibrotic liver [48]. The abnormal lipid metabolism in liver mostly results in inflammation and liver fibrosis [49]. Mice fed a high-fat diet experiences cardiac fibrosis even before obesity and hyperlipidemia develops [50]. Inflammatory lipid mediators such as prostaglandins, leukotrienes, and epoxyeicosatrienoic acids might be involved in cardiac fibrosis [51]. Changes in bioactive phospholipids such as phosphatidic acid and lysophosphatidic acid, compounds involved in cell proliferation, migration, and survival, were observed in the bronchoalveolar lavage of patients suffering from idiopathic pulmonary fibrosis [52]. Branched amino acids have been reported to reduce liver fibrosis via transforming growth factor β inhibition [53]. These studies indicated that a metabolomics study would be a promising strategy for identifying prognostic or diagnostic biomarkers of fibrosis and for better understanding of disease mechanisms.
Most cancer cells predominantly produce energy for rapid cell proliferation via highly activated glycolysis that is characterized by lactic acid accumulation. This phenomenon of cancer cells, also known as the Warburg effect, indicates an adaptation to the low oxygen environments within tumors or damage of mitochondria in cancer [54]. Metabolic reprogramming in cancer has attracted attention as a new avenue for cancer research [555657]. Metabolic alterations in cancer might include numerous metabolic pathways affecting cell growth and proliferation, including those of energy metabolism, lipid metabolism, and nucleotide metabolism [58]. Bioactive lipids such as eicosanoids are linked to cancer. The roles of these bioactive lipids in the interactions between transformed epithelial cells and the surrounding stromal cells are crucial for understanding tumor evolution, progression, and metastasis [59]. However, the roles of lipids in tumors have been poorly elucidated. The global lipid profiles of the breast cancer tissues of 267 human subjects showed that de novo fatty acid synthesis was higher in tumors than in normal breast tissues, possibly for the incorporation of fatty acids into membrane phospholipids. Immunohistochemical analysis of the expression of specific proteins, selected from in silico transcriptome database searches, showed that lipid metabolism related to de novo fatty acid synthesis was highly expressed [60]. Further understanding of the molecular mechanisms underlying the roles of metabolites in cancer progression should be useful for the development of more effective targets for cancer therapies.
CONCLUSIONS
Metabolomics is a relatively new omics strategy in biomedical research and is gaining attention as a tool for the discovery of biomarkers for disease diagnosis and assessment of disease development and prognosis. In addition, metabolomics may provide evidence for the identification of candidates for novel therapeutic interventions and the elucidation of disease mechanisms. The successful application of metabolomics requires integrated efforts from analytical chemistry, biology, medicine, and biostatistics.



 XML Download
XML Download