

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
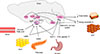
Over the past decades, our knowledge of the neuronal regulation of energy homeostasis has dramatically expanded. Substantial evidence indicates that the brain plays a central role in the homeostatic regulation of energy metabolism. The brain integrates multiple peripheral metabolic inputs, such as nutrients, gut-derived hormones, and adiposity-related signals. This information on energy intake and body energy stores is transferred to specialized neurons in the hypothalamus and brainstem. In order to maintain energy homeostasis, the brain regulates diverse aspects of body metabolism, such as food-seeking behavior; gastric emptying; nutrient uptake in the gut; thermogenesis; pancreatic insulin secretion; and the effects of insulin in the liver, adipose tissue, and skeletal muscle. In this review, we describe the brain's regulatory mechanisms of food intake and energy expenditure (Fig. 1).
BRAIN REGULATION OF FOOD INTAKE
The hypothalamus is the region of the brain that controls food intake and body weight. The hypothalamic arcuate nucleus (ARC) is ideally situated near the third ventricle and the median eminence, which is an area with a relatively porous blood-brain barrier. This provides the ARC free access to circulating nutrients and hormones, making it the primary nutrient-sensing center of the hypothalamus. There are two distinct neuronal populations in the ARC, orexigenic neurons that express both neuropeptide Y (NPY) and agouti-related peptide (AgRP) and anorexigenic neurons that express proopiomelanocortin (POMC). These neurons are first-order neurons that respond to peripheral metabolic signals and project to second-order neurons of the paraventricular nucleus (PVN), the perifornical area adjacent to the fornix and the lateral hypothalamus (LH), and to autonomic preganglionic neurons in the brain stem and spinal cord.
POMC neurons produce the anorectic peptide α-melanocyte stimulating hormone (α-MSH) by posttranscriptional processing of POMC. α-MSH binds to the melanocortin receptors 3 and 4 (MC3R and MC4R) on second-order neurons and activates catabolic pathways, leading to reduced food intake and increased energy expenditure [1]. On the other hand, central administration of NPY increases food intake via Y1 or Y5 receptors, which are highly expressed in the ARC, PVN, and ventromedial hypothalamus (VMH) [2]. Likewise, central administration of AgRP induces hyperphagia and weight gain by inhibiting the binding of α-MSH to MC3R/MC4R [3]. Selective ablation of NPY/AgRP neurons in adult mice results in anorexia and weight loss [4], demonstrating a critical role of these neurons in the regulation of energy homeostasis.
Both POMC and NPY/AgRP neurons in the ARC alter their activity in response to blood glucose level [5]. Elevated extracellular glucose level activates POMC neurons, whereas NPY/AgRP neurons are activated in glucose-deprived conditions. Hypothalamic neuronal glucose deprivation induced by administration of 2-deoxy-D-glucose potentially increases food intake [6]. Circulating long-chain fatty acids (LCFAs) also act as nutrient abundance signals in the hypothalamus. Intracerebroventricular administration of LCFAs, specifically oleic acid, inhibits food intake by decreasing hypothalamic AgRP and NPY expression [7]. Increased levels of lipid metabolites such as malonyl CoA and LCFA-CoA in hypothalamic neurons are indicative of nutrient excess and lead to signals for food intake reduction [78]. The hypothalamic ARC is critical for sensing adiposity signals such as leptin and insulin. Leptin and insulin signal the status of body energy stores to the hypothalamus. Both leptin and insulin activate POMC neurons, while inhibiting NPY/AgRP neurons [910]. Ghrelin is a gut hormone secreted from the stomach during fasting, signaling the need for increased food intake. The orexigenic action of ghrelin is mediated by NPY/AgRP neurons in the ARC [11].
The PVN is an important brain region that regulates neuroendocrine function by releasing critical neuropeptides, including oxytocin, thyrotropin-releasing hormone, and corticotrophin-releasing hormone. The PVN is also important for regulating energy balance. The PVN neurons are densely innervated by POMC and NPY/AgRP neurons [12], and they serve as secondorder neurons in the melanocortinergic neuronal circuit. For example, PVN oxytocin neurons mediate melanocortinergic control of food intake by innervating and regulating neurons in the nucleus of the solitary tract (NTS) [13]. Moreover, the PVN is a key brain region that mediates the actions of glucagon-like peptide-1 (GLP-1), an important gut-derived satiety signal [14].
The VMH neurons mainly receive neuronal inputs from the ARC and then project their axons to the ARC, PVN, LH, dorsomedial nucleus (DMN), and the NTS. Most VMH neurons express steroidogenic factor 1 (SF-1) [15], and those VMH neurons that express SF-1 release brain-derived neurotrophic factor (BDNF). Selective deletion of BDNF in the VMH results in hyperphagia and obesity in mice [16]. Moreover, loss of function mutations in both BDNF and the BDNF receptor tropomyosin receptor kinase B cause hyperphagia and severe obesity in humans and in rodents, indicating that BDNF is an important satiety factor [1718].
The LH is in the hypothalamic region where metabolic and reward-related information is integrated. This information is transferred to various brain areas such as the hindbrain, cortex, limbic system, thalamus, and spinal cord, allowing for a complex modulation of both behavioral and autonomic outflow. The LH contains two distinct neuronal populations that produce melaninconcentrating hormone (MCH) and orexin, respectively. Orexin demonstrates appetite-enhancing actions of directly activating NPY/AgRP neurons and indirectly inhibiting POMC neurons in the ARC [1920]. MCH also exerts orexigenic effects by modulating the ARC melanocortin system [21]. Genetic overexpression of MCH in mice leads to hyperphagia and obesity [22].
The brainstem is another major brain area involved in the control of food intake. Meal-elicited gastrointestinal signals induce neuronal activation in the caudal brainstem, where vagal afferents terminate. Given that chemical and surgical vagal denervation is known to decrease meal size and duration, it is thought that meal-related signals are transferred to the brain via the vagal afferent [23]. The NTS is a major neuronal connection between the gut and brain. Like the ARC, the NTS is anatomically close to the area postrema. Thus, the NTS is specialized for receiving both humoral and neural signals from the periphery. Extensive reciprocal neuronal connections exist between the hypothalamus and the brainstem, and the amount of food intake is determined based on metabolic information delivered to both brain regions [24]. Metabolic signals from gut hormones are transferred to the brainstem through the vagal nerve. Cholecystokinin, GLP-1, and peptide YY are released from the enteroendocrine cells upon food intake, and they bind their receptors on the vagus nerve terminals. These food intake signals are delivered to the hypothalamus via the NTS; thereby, inducing satiety [2526]. Like hypothalamic neurons, NTS neurons produce POMC, NPY, and GLP-1. POMC-producing NTS neurons are activated upon food intake. These neurons also exhibit signal transducer and activator of transcription 3 (STAT3) activation in response to exogenous leptin [27], suggesting a role of brainstem neurons in sensing peripheral metabolic signals.
BRAIN REGULATION OF ENERGY EXPENDITURE
Energy is consumed in the processes of physical activity, basal metabolism, and adaptive thermogenesis, all of which are modulated by the brain. The hypothalamic ARC is considered a key site for mediating leptin's effect on locomotor activity, since selective restoration of leptin signaling in the ARC, especially in POMC neurons, normalized locomotor activity in leptin receptor-null mice [28]. Meanwhile, NPY, AgRP, and orexin promote food-seeking behavior [2930].
Thermogenesis refers to heat that is generated in order to maintain body temperature or in order to dissipate excess energy upon food intake. Brown adipose tissue (BAT), located in the interscapular area of rodents, plays a major role in thermogenesis [31]. Thermogenesis is a critical component of energy expenditure, especially in rodents. Central regulation of BAT thermogenesis is dependent on sympathetic outflow to BAT. Norepinephrine released from sympathetic nerve terminals binds to β3-adrenergic receptors on adipocytes in BAT and inguinal fat pads. Activated adrenergic receptors trigger cyclic-adenosine monophosphate signaling, which activates mitochondrial uncoupling protein-1 (UCP-1) and promotes enhanced thermogenesis.
The preoptic area (POA) has been identified as the neural circuit that regulates sympathetic outflow to BAT by directly projecting to sympathetic premotor neurons in the rostral raphe pallidus [32]. Alternatively, POA neurons project to DMN neurons, which mediate POA-evoked thermoregulatory responses [33]. In addition, many hormonal and nutrient signals, such as glucose, insulin, leptin, and GLP-1, can influence sympathetic outflow to BAT [3435]. Central administration of an MC3R/MC4R agonist stimulates BAT activity through sympathetic outflow, suggesting a regulatory role of the hypothalamic melanocortin system in BAT thermogenesis [36].
Brown-like fat adipocytes, so-called “beige” or “brite” adipocytes, are found in the inguinal subcutaneous area of rodents and in the supraclavicular, suprarenal, pericardial, and para-aortic areas and around the pancreas, kidney, and trachea in humans [37]. UCP-1 expression in beige adipocytes is low under basal conditions, but its expression is induced under certain circumstances, such as exposure to cold temperatures. Induction of white adipose tissue (WAT) browning in rodents increases energy expenditure and attenuates diet-induced obesity [38]. Conversely, blockade of WAT browning through deletion of Prdm16, a transcriptional coregulator that controls the development of brown adipocytes, promotes obesity [39]. Interestingly, insulin and leptin act synergistically on POMC neurons to promote both WAT browning and energy expenditure. These mechanisms seem to be important for resistance against the development of diet-induced obesity [40]. Therefore, it is thought that POMC neurons convey leptin and insulin signaling to drive WAT browning and to enhance energy expenditure.
CONCLUSIONS
Obesity has reached epidemic levels worldwide, accompanied by the increased prevalence of multiple comorbidities. Considerable attention is now being paid to understand how our body maintains energy balance under healthy conditions and why these mechanisms become defective in obesity and cachexia. Expanding our knowledge of the brain's regulation of food intake and energy expenditure will lead us to effective therapeutic strategies for combating obesity and related metabolic disorders.
 XML Download
XML Download