

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
A large number of studies has clearly shown the existence of a strong inverse correlation between plasma high-density lipoprotein cholesterol (HDL-C) concentrations and the incidence of coronary heart disease (CHD) [12], but the significance of such association has been recently questioned. Intervention clinical trials carried out with agents efficient in raising HDL-C levels, including niacin and cholesteryl ester transfer protein (CETP) inhibitors, have failed in showing a reduction in cardiovascular events [345]. In addition, Mendelian randomization studies have shown that increased HDL-C levels caused by common variants in HDL related genes are not necessarily associated with reduced cardiovascular risk [678]. One possible explanation for this discrepancy is that the plasma HDL-C concentration does not reflect the very complex HDL system, involving different HDL particles and a number of receptors, transporters, enzyme, and transfer proteins [9]. Moreover, cholesterol is not the active component of HDL and there is convincing evidence that at least some of the atheroprotective functions of HDL relate to specific HDL components or subclasses, which concentration in plasma may be totally unrelated to the HDL-C level [1011].
HDL METABOLISM AND FUNCTION
HDL heterogeneity
HDL are a highly heterogeneous lipoprotein family composed by several subclasses with different density, shape, and size. The density of the HDL particles is inversely related to their size, reflecting the relative contents of low density non-polar core lipid, and high density surface protein. Most part of plasma HDL has a globular shape, the central core is composed by non-polar lipids (triglycerides and cholesteryl esters) surrounded by a monolayer of polar lipids (phospholipids and unesterified cholesterol) and apolipoproteins [12]. A minor fraction of plasma HDL has a non-spherical structure and they consist in discoidal bilayer of polar lipids, in which non-polar core is lacking; apolipoproteins run from side to side of the disk, with polar residues facing the aqueous phase and non-polar residues facing the acyl chains of the lipid bilayer [13]. The protein component of HDL is formed mainly by apolipoprotein A-I (apoA-I), for the 70%, and apolipoprotein A-II (apoA-II), for the 20%. Two major particle subclasses have been identified on the basis of major apolipoprotein composition: particles containing only apoA-I (LpA-I), and particles containing both apoA-I and apoA-II (LpA-I:A-II). Recent shotgun proteomic analysis showed that HDL contain 48 or more proteins, among these apoA-IV, apoCs, apoE, lecithin:cholesterol acyltransferase (LCAT), CETP, phospholipid transfer protein (PLTP), paraoxonase (PON), and platelet-activating factor acetylhydrolase (PAF-AH) circulate in plasma bound to HDL [91214]. Most of the proteins carried by HDL are not apolipoproteins, and represent very minor components of these particles.
It is also possible to classify HDL on the basis of density (HDL2, with density of 1.063 to 1.120 g/mL, and HDL3, 1.120 to 1.210 g/mL), charge, shape, and size. According to charge, HDL can be divided into α- and pre-β-migrating particles on agarose gel, and combining charge and size these two subclass-es can be divided into 12 distinct apoA-I-containing particles, referred to as pre-β (pre-β1 and pre-β2), α (α1, α2, and α3) and pre-α (pre-α1, pre-α2, and pre-α3) on the basis of mobility that is slower or faster than albumin, respectively, and decreasing size [15].
HDL metabolism
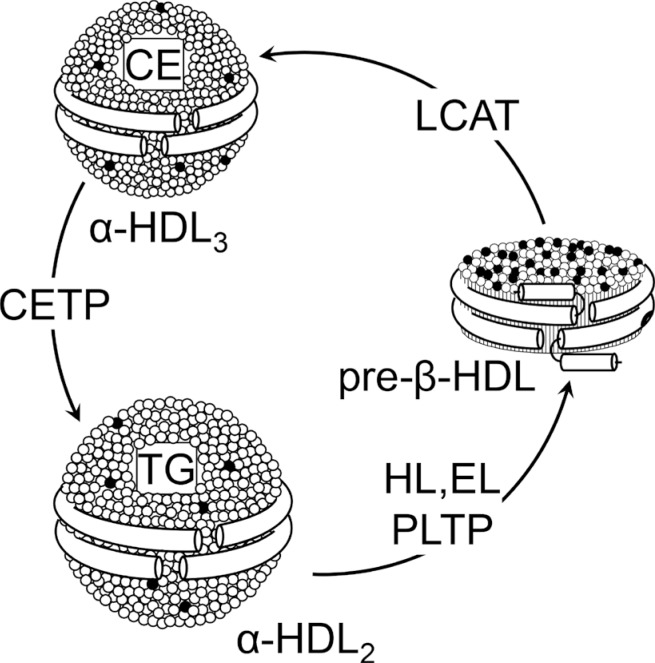
Due to its highly dynamic nature, apoA-I is involved in major pathways of HDL metabolism. ApoA-I and apoA-II are synthe-sized mainly by the liver and, to a lesser extent, by the small intestine [16] and are secreted as components of triglyceride-rich lipoproteins. In circulation, PLTP promotes the transfer of surface components (phospholipids, cholesterol, and apolipoproteins) from triglyceride-rich lipoproteins to HDL. The regulatory role of PLTP is achieved through two main functions, phospholipid transfer activity and the capability to modulate HDL size and composition in a process called HDL conversion [17]. Hepatocytes are able to secrete apoA-I in lipid-free or lipid-poor and lipidated forms [18]. ApoA-I is secreted as pro-apoA-I and converted to a mature form by a metalloprotease in plasma [19]. There are three potential sources of lipid-poor apoA-I in plasma: it may be released as lipid-poor protein after its synthesis in the liver and intestine, it may be released from triglyceride-rich lipoproteins that are undergoing lipolysis by lipoprotein lipase, and it may be generated in the circulation during the remodeling of mature, spherical HDL particles. Lipid free-apoA-I acquires phospholipids and cholesterol through the interaction with the ATP binding cassette transporter A1 (ABCA1), to form pre-β-HDL, a pathway dependent on ABCA1 expression [20]. Once in the circulation, pre-β-HDL are the preferential substrate of LCAT (Fig. 1), that converts lecithin and cholesterol into lysolecithin and cholesteryl esters, using apoA-I as cofactor [21]. The cholesterol esters generated by LCAT are more hydrophobic than free cholesterol and thus migrate into the hydrophobic core of the lipoprotein, with the resulting conversion of small, discoidal pre-β-HDL into mature, spherical, α-migrating HDL (α-HDL) [21]. LCAT thus plays a central role in intravascular HDL metabolism and in the determination of plasma HDL level. Esterification of cholesterol in plasma by LCAT is also necessary for cholesterol uptake from the liver, either directly through the scavenger receptor class B member 1 (SR-BI) or indirectly through CETP. The α-HDL produced by LCAT (HDL3) interact in the plasma with CETP, that exchanges cholesteryl esters for triglycerides between HDL and triglyceride-rich lipoproteins, generating large cholesteryl ester-poor and triglyceride-rich HDL particles (HDL2) [22]. Mature, large α-HDL particles can be converted back to pre-β-HDL through the action of PLTP and the endothelial and hepatic lipases, that hydrolyze triglycerides and phospholipids on HDL (Fig. 1) [23]. Plasma half-life of pre-β-HDL is short, they are rapidly cleared through the kidney, while mature α-HDL have a slower turnover [24].
HDL components are catabolized in different ways; the major sites of catabolism of the protein components are liver and kidney. The kidney, according to hydrophobicity, filters lipid-free apolipoproteins; apoA-I and apoA-II can be reabsorbed through cubulin receptors in the kidney proximal tubules [25]. When reabsorption is impaired, hydrophilic apolipoproteins (apoA-I and apoA-IV, but no apoA-II) can be excreted into urine [26]. Glomerular filtration barrier prevents access of mature HDL particles to the proximal tubules; however, cubulin may bind filtered lipid-poor HDL [27]. HDL particles can entirely be removed by holoparticles HDL receptors. In the liver, holo-HDL particles accumulated in endosomal compartments can be transferred to lysosomes for degradation or in a small proportion, they can be resecreted into the circulation [28].
HDL functions
One of the most important function of HDL is to promote the removal of cholesterol from peripheral cells, including macrophages within the arterial wall, and shuttle it to the liver for excretion through the bile and feces in a process called reverse cholesterol transport (RCT). It results in a net mass transport of cholesterol from the arterial wall into the bile. This pathway is described as an anti-atherogenic process by preventing arterial cholesterol accumulation, plaque destabilization, and development of acute cardiovascular events [9]. Cell cholesterol efflux is the first and limiting step in RCT and consists in the exchange of unesterified cholesterol between cells and extracellular acceptors [29]. This exchange can occur by several processes: via aqueous diffusion, which occurs according to the direction of cholesterol gradient, or through three main distinct and protein-mediated pathways [29]. The various HDL particles are differently efficient in promoting cell cholesterol efflux. Lipid-free/lipid-poor apolipoproteins, mainly apoA-I, represent the principal cholesterol acceptors via ABCA1 [29]. All plasma HDL subclasses, including mature α-HDL particles and discoidal pre-β-HDL, are efficient cholesterol acceptors via the ABCG1 pathway, while SR-BI promotes cell cholesterol efflux only to mature, large α-HDL [30]. The cholesterol accumulated in HDL is esterified in plasma by LCAT with the resulting formation of cholesteryl esters. Then, hydrophobic cholesteryl esters move to the core while unesterified cholesterol is removed from the surface of HDL, leading to the progressive enlargement of these particles. For long time, LCAT has been considered necessary for efficient RCT by keeping unesterified cholesterol gradient from cells to HDL, but recent data suggest that even if functional LCAT is not present, macrophage cholesterol efflux and RCT can occur [31]. Large amount of the cholesteryl esters formed by the LCAT are exchanged with triglycerides through CETP-mediated process into apoB containing lipoproteins that are finally catabolized by the liver. Alternatively, HDL-cholesteryl esters are taken-up by the liver through SR-BI [32].
Atheroprotection mediated by HDL is not only through their major role in RCT, but also through other relevant functions; one of the most important and well studied is the ability of HDL to maintain endothelial cell homeostasis and integrity [33]. HDL have potent antioxidant properties, mediated by molecules carried by HDL (PON-1, PAF-AH, and LCAT) or by apoA-I and apoA-II, as well as anti-inflammatory, antithrombotic, cytoprotective, vasodilatory, anti-infectious activities and the capacity to enhance insulin secretion. This wide spectrum of biological activities likely reflects the heterogeneity of HDL particles; however, the HDL-protective activities can be lost in some pathological conditions and HDL can even acquire proatherogenic properties [34].
LCAT AND ATHEROSCLEROSIS
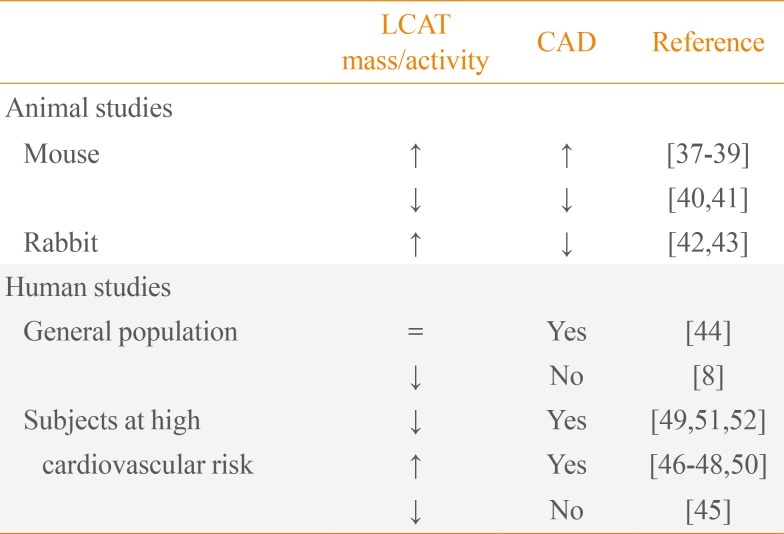
LCAT is principally synthetized in the liver and in little amount in other tissues, such as brain and testes, and circulates in plasma compartment at concentration of 5 mg/L mainly bound to HDL but also to LDL. LCAT converts phosphatidylcholine and cholesterol into cholesteryl ester and lysophosphatidylcholine in plasma and other biological fluids [21]. In RCT pathway, LCAT plays a key role and it is thought to help facilitate this process by leading formation of large and mature HDL. Furthermore, it is reported that the majority of cholesteryl esters formed by LCAT are removed by the liver [35]. Without LCAT, HDL-C, apoA-I and apoA-II levels in the plasma are drastically reduced for the lacking formation of mature and spherical shaped HDL and for the rapid catabolism of discoidal HDL by the kidney [36]. On the basis of these evidences, variations in LCAT activity seem to be naturally implicated in atherosclerosis prevention or development. To elucidate the role of LCAT in atherosclerosis, a large number of studies have been performed in both animal models and humans (Table 1) [837383940414243444546474849505152].
Studies carried out in animal models led to controversial results, often dependent on species utilized. Studies performed in mice in which LCAT was overexpressed [373839] or downregulated [4041] suggest that activity of LCAT is not associated to atheroprotection and the lack of enzyme is not associated to increased atherosclerosis, even if the HDL-C levels in plasma are very low. The increased atherosclerosis in mice with LCAT overexpression is probably due to the accumulation in plasma of dysfunctional large apoE-rich HDL, which were shown to be defective in the delivery of cholesterol to the liver through SR-BI [39]. When the LCAT gene was overexpressed in rabbits, opposite results were obtained: aortic lesions were reduced after atherogenic diet, even if large HDL particles containing apoE were detected [4243]. The contradictory results obtained in the studies on animal models do not clarify the role of LCAT in atherosclerosis, allowing for further consideration.
The role of LCAT in atherosclerosis was also explored in humans, both in general population and in subjects at high cardiovascular risk. As observed in animal studies, the role of LCAT in the pathogenesis of human atherosclerosis remains controversial. The Epic-Norfolk was the first prospective study investigating the correlation of LCAT plasma levels and atherosclerosis carried out in general population in more than 2,700 subjects [44]. One-third of enrolled subjects developed coronary artery diseases (CADs), but no associations between plasma LCAT levels and risk to develop future CAD was observed. When individuals were divided according to gender, increased LCAT levels correlated with lower risk of CAD only in men, while in women was the opposite [44]. Reduction of LCAT concentration/activity associated with absence of CAD was described in The Copenhagen City Heart Study, that enrolled more than 10,000 participants, and in The Copenhagen General Population Study, in which more than 50,000 subjects are involved [8]. The variants S208T found in the coding region of LCAT gene was associated with reduction in HDL-C and apoA-I levels, but not with increased risk of myocardial infarction, ischemic heart disease, and ischemic cerebrovascular disease [8]. In agreement with the results obtained in the general population, an observational study carried out in 540 subjects at high cardiovascular risk showed that low plasma LCAT levels are not associated with higher carotid intima-media thickness (IMT) [45], a marker of preclinical atherosclerosis.
Consistent with these results, in various studies it was demonstrated that an increased LCAT concentration is associated to CAD. Increased levels of LCAT activity was associated with increased IMT in 74 subjects with metabolic syndrome [46], as well as in the control subjects of the study [46]. In another study from the same group, the association between LCAT activity and CAD was found only in men [47]. A recent study analyzed the relationship between LCAT activity and triglyceride metabolism and LDL particle size in 550 patients at high cardiovascular risk [48]. Increased LCAT activity was associated with formation of small LDL particles that are more atherogenic than large particles, but no parameters of subclinical atherosclerosis were analyzed [48].
On other side, some studies affirm the opposite: decreased LCAT activity is associated with CAD. Early studies supporting this evidence were carried out in 1973 in subjects at high cardiovascular risk [49]. Few years later, in 100 subjects divided according to the degree of atherosclerotic disease, LCAT activity was found positively correlated with the severity of coronary atherosclerosis [50]. Lower levels of LCAT activity were also observed in patients with ischemic heart disease [51], and in a study on patients with acute myocardial infarction [52].
CONCLUSIONS
While epidemiological studies have repeatedly shown a strong and inverse correlation between plasma HDL-C concentrations and the incidence of CHD, the significance of such association for CHD development has been recently questioned, and clinical trials with various drugs able to increase HDL-C levels did not show the expected benefits. HDL metabolism is regulated by a large number of factors that modify plasma levels of circulating HDL, and plasma HDL-C levels are remarkably susceptible to variations in these factors which also affect HDL shape, size, density, and lipid and apolipoprotein composition, and as a consequence HDL function. Investigations of factors involved in HDL metabolism thus represent a good way to understand the relationship between HDL and CHD, and will likely translate in the development of innovative therapeutic approaches to CHD prevention and treatment specifically affecting HDL function independent of plasma HDL-C levels.
 XML Download
XML Download