

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) are incretin hormones that potentiate glucose-stimulated insulin secretion from β-cells [1]. Recently, new therapeutic agents such as GLP-1 analogs and dipeptidyl peptidase-4 (DPP4) inhibitors were introduced to clinical practice with proven efficacy of glucose control [1]. Development of these novel medications was based on a marked reduction of incretin effects in patients with type 2 diabetes mellitus (T2DM) [2]. In patients with T2DM, the reduction of incretin effects was largely due to decreased GLP-1 secretion after nutrient stimulation or ineffective GIP action [3]. Nevertheless, it has been demonstrated recently that the incretin effect is not impaired in Japanese and Korean T2DM subjects [45]. Recent meta-analyses of clinical studies also suggested that patients with T2DM, in general, do not exhibit reduced GLP-1 secretion in response to an oral glucose tolerance test (OGTT) or meal test [6]. There are ethnic differences in the pathogenesis of T2DM, especially in Caucasian and East Asians [7]. However, studies of incretin action in East Asians are limited.
It is well established that the risk of diabetes in prediabetic subjects is much higher compared to those with normal glucose tolerance (NGT) [8]. Even among subjects with NGT, an upper normal level of fasting plasma glucose (FPG) is a predictor of T2DM [9]. However, incretin hormone levels as a predictor of T2DM have not been fully investigated because incretin levels have only been measured in small groups of subjects [1011] due to the relatively laborious and time-consuming nature of the laboratory methods associated with this measurement. The present study was conducted to evaluate incretin hormone levels before diabetes develops and to determine the role of incretin hormones as predictors of diabetes development.
METHODS
Study design and participants
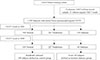
A nested case-control analysis was performed using participants of an Ansung cohort study. The design and baseline characteristics of the Ansung-Ansan cohort study have been described in detail elsewhere [12]. Briefly, it is an ongoing prospective community-based epidemiological study that is a part of the Korean Health and Genome Study which was conducted to investigate trends in diabetes and associated risk factors. The baseline examination was performed in 2001 to 2002, and biennial follow-up examinations were continued through 2012. Of the 5,018 subjects who were surveyed in Ansung, complete data from the baseline investigation and frozen samples for further analysis were available for 1,371 participants who registered for the cohort study in the first year (2001). Among the 1,371 subjects, the results of OGTT were available for 1,358 subjects. During both initial screening and follow-up visit, the definitions of NGT, prediabetes and diabetes were based on plasma glucose levels during the 75 g OGTT according to the 1997 American Diabetes Association criteria. NGT was defined as FPG level <6.1 mmol/L and 2-hour plasma glucose <7.8 mmol/L. Prediabetes was defined as 6.1 mmol/L≤FPG level<7.0 mmol/L or 7.8 mmol/L≤2-hour plasma glucose<11.1 mmol/L. Diabetes was defined as FPG concentration ≥7.0 mmol/L or 2-hour plasma glucose ≥11.1 mmol/L or current treatment with oral antidiabetic drugs or insulin [13]. At the time of initial screening in 2001, there were 948 subjects with NGT, and 261 subjects with prediabetes. Among the subjects who showed NGT in 2001, 23.9% (227/948) developed impaired fasting glucose or impaired glucose tolerance (IGT) and 6.9% (65/948) developed diabetes by 2009. The "incident diabetes group (NGT→diabetes mellitus group)" in the present study consisted of the 65 subjects who developed diabetes during the follow-up period. Among the 656 subjects who maintained NGT during the 8-year follow-up period, 149 subjects were randomly selected and matched to diabetic subjects by sex and age. These 149 subjects were defined as the "control group (NGT→NGT group)" (Fig. 1). Circulating levels of total GLP-1 and total GIP were measured in selected samples to examine the relationship between basal incretin hormones and diabetes development. The study protocol was approved by the Institutional Review Boards of the Samsung Medical Center (Approval number 2010-11-068-001).
Data collection and biochemical analyses
Anthropometric parameters and blood pressure were measured using standard methods. FPG, insulin, total cholesterol, triglycerides, and high density lipoprotein cholesterol were measured, and the results were obtained from the main database. Homeostasis model of assessment-insulin resistance (HOMA-IR) was defined as [fasting insulin (µU/mL)×fasting glucose (mmol/L)]/22.5. Homeostasis model of assessment-β cell function (HOMA-B) was calculated using (20×fasting insulin in µU/mL)/(fasting glucose in mmol/L-3.5) [14]. Total GLP-1 and total GIP were measured in the stored samples. The plasma concentrations of total GLP-1 (ALPCO Diagnostics, Windham, NH, USA) and total GIP (Millipore Corp., Bedford, MA, USA) were measured without an ethanol extraction step by an enzyme-linked immunosorbent assay. Samples were frozen at -70℃ and never thawed until they were moved to Samsung Medical Center for analyses. The biochemical analysis of incretin hormones was performed in duplicate by a single trained technician in the Department of Laboratory Medicine and Genetics at the Samsung Medical Center.
Statistical analyses
Statistical analyses to compare baseline characteristics between and among the groups were conducted using one-way analysis of variance for normally distributed data. The Kruskal-Wallis test was used for analysis of skewed data. Categorical variables were compared using Pearson and chi-square test. Univariate analyses were performed to assess the association between diabetes development and clinical parameters. Multivariate analysis was performed using variables from the univariate analyses that were significant at P<0.05. A binary logistic regression model was used for univariate and multivariate analyses. P<0.05 was considered statistically significant. A binary logistic regression model was used for development of diabetes according to incretin quintiles. Statistical analyses were performed using IBM SPSS version 19 (IBM Co., Armonk, NY, USA).
RESULTS
Descriptive data
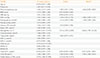
Demographic characteristics and laboratory results are summarized in Table 1. Demographic characteristics such as age and sex were not significantly different between groups. On the other hand, significant differences in waist circumference, body mass index (BMI), systolic blood pressure, diastolic blood pressure, FPG, postprandial glucose, hemoglobin A1c (HbA1c), triglyceride, aspartate transaminase, alanine transaminase (ALT), γ-glutamyl transpeptidase were observed. Those discrepancies were primarily observed in subjects who had diabetes upon initial enrollment. To investigate the role of incretin hormones as predictors of incident diabetes, total GLP-1 and total GIP levels were compared between patients who developed diabetes and those who maintained NGT. Total GLP-1 levels were not significantly different between the two groups who developed diabetes and those who maintained NGT (P=0.199). Nonetheless, there were significant differences in total GIP levels between patients who developed diabetes and those who did not (P=0.034). Correlations between incretin hormones and HOMA-B and HOMA-IR were not significant (data not shown).
Risk estimates for diabetes development
Parameters that were significantly associated with diabetes development included waist circumference, BMI, blood pressure, FPG, postprandial glucose, HbA1c, triglycerides, family history of diabetes, and total GIP levels (Table 2). In multivariate analyses, incretin levels were not significantly associated with the development of diabetes (model 1 of Table 2). After selecting the most relevant variables to predict diabetes occurrence, a second model was established (model 2 of Table 2). In this second model, fasting total GIP levels were significantly associated with diabetes development, whereas GLP-1 levels were not. In both univariate and multivariate analyses, higher HbA1c was the most significant predictor of diabetes development. In multivariable logistic regression analyses adjusted for age, sex and BMI, total GIP levels in the fifth quintile were associated with higher odds ratios of future diabetes development, while total GLP-1 levels were not. This association remained significant after additional adjustment for HbA1c (Table 3).
DISCUSSION
The purpose of this study was to investigate the association between incretin hormones and diabetes development in a community-based cohort. The novel finding is that fasting GIP levels were elevated before diabetes development even in subjects with NGT. In patients with T2DM, the reduced effects of incretin are well elucidated [2]. Reduced incretin effects have been explained by diminished GLP-1 levels in plasma and deteriorated GIP action [3]. Although impaired GLP-1 secretion after nutritional stimuli in diabetic patients has been reported in several papers [1516], GLP-1 secretion is currently believed to be preserved in diabetic patients [6]. Some researchers have proposed that reduced incretin effect is a consequence of diabetic status, as deterioration in glucose homeostasis can develop in the absence of any impairment in GIP or GLP-1 levels [10]. The findings of a large cohort study demonstrated that fasting and integrated postprandial concentrations of total and active GLP-1 are not significantly different between subjects with NGT and those with IGT [17]. In addition, recent case-control study also found no difference in fasting GLP-1 among normal, prediabetes and diabetes groups [18]. The present study had similar findings when comparing GLP-1 levels in normal and diabetic subjects on enrollment (data not shown).
GIP was the first incretin identified. In addition to stimulating insulin secretion, GIP plays regulatory roles in the maintenance, growth and survival of pancreatic islets, as well as impacting on adipocyte function [19]. In the present study, GIP levels were increased significantly in subjects who later developed diabetes. This result is also in line with previous report showing that GIP levels were significantly elevated in newly-diagnosed diabetes group when compared with the normal group [18]. The mechanisms for these findings are unclear, but GIP elevations might be caused by GIP receptor resistance or defective GIP receptor expression. GIP receptor gene polymorphism was shown to be associated with diabetes and the metabolic syndrome, and GIP receptor polymorphism has begun to receive more attention than GIP plasma levels [20]. GIP/GIP receptor axis is also known to be disrupted in insulin-resistant states, such as obesity [21]. A meta-analysis of nine genome-wide association studies was conducted in order to identify T2DM-associated loci, and it was concluded that genetic variation in the GIP receptor influences glucose and insulin responses to an oral glucose challenge [22]. Most recent study found that up-regulation of GIP production through interaction with GIP receptors on islets may be a key factor in multi-hormonal dysregulation in T2DM [18]. Additionally, similar findings were reported in various animal models [232425]. Our results are in accordance with many studies [26] supporting the role of GIP in an early pathophysiological step that could lead to T2DM. GIP secretion was also known to be preserved in response to OGTT or meal test in patients with T2DM [27], but the effect of GIP on insulin is blunted [28]. Collectively, increased GIP levels may lead to development of T2DM in similar way like insulin resistance.
However, GIP level in subjects with diabetes was not different from the GIP level of the subjects who remained normal during follow-up period. GIP level decrement after diabetes development is hard to explain because little is known about the molecular mechanism of GIP secretion, although several factors which are associated with GIP gene expression have been reported [2930]. Recent experimental study with T2DM patients found that GIP has negligible effect on plasma glucose at fasting glycemia but it retains insulinotropic effects only during hyperglycemia [31]. Therefore, full GIP response data after meal is further needed to explain this result. Similar patterns are also seen in C-peptide changes during T2DM development. In the early of stages of T2DM, C-peptide levels are usually elevated compared to normal subjects; however, as the duration of T2DM becomes longer, C-peptide levels decrease [32]. ALT was associated with increase in the risk of T2DM even if it is not elevated in the subjects with diabetes [12]. Another explanation is that GIP level can be affected by anti-diabetic drugs. We could not adjust the effect of glucose lowering medication on the serum incretin level in the diabetes group due to lack of data. However, incretin mimetics were not available in the year of 2001 to 2002. Further investigations are required to clarify the causal relationships between the GIP level and diabetes development.
To the best of our knowledge, this is the first study that measured incretin levels in a large Asian community-based cohort. Moreover, this was an 8-year prospective study of the relationship between incretin hormones and diabetes development. The most significant limitation is that the samples were fasting samples, and thus it was impossible to determine the response of incretin hormones to meal stimuli. However, it seems reasonable to assume that fasting total GIP levels might be used to estimate enteroinsular axis like the homeostasis model assessment. In homeostasis model assessment, FPG and insulin levels are used to estimate insulin resistance and β-cell function [14]. Another limitation is that the samples were not preserved in DPP4 inhibitor containing tube.
In summary, elevated GIP levels were associated with increased diabetes risk in this study. After adjusting for other associated risk factors, fasting GIP levels might be a risk factor for the development of diabetes mellitus, suggesting that deterioration in the enteroinsular axis might occur before diabetes develops.


 XML Download
XML Download