

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Allgrove for the first time in 1978 described two pairs of siblings with adrenal insufficiency, achalasia cardia and alacrimia [1]. The combination of these three manifestations was known as Allgrove syndrome or Triple A syndrome. The spectrum of disease varies widely and sometimes it is expressed by only two of the three cardinal manifestations. Neurological dysfunction from the involvement of central, peripheral or autonomic nervous systems is often associated with Allgrove syndrome [2]. Adrenal insufficiency and achalasia are usually evident by the end of first decade and neurological dysfunction follows later.
Triple A syndrome is rare and has been reported from different parts of the world. In the literature it has been described as case reports. The molecular basis of this rare autosomal recessive disorder is the mutated achalasia, adrenocortical insufficiency, alacrimia syndrome (AAAS) gene, located on chromosome 12q13, that codes for ALacrima Achalasia aDrenal Insufficiency Neurologic disorder (ALADIN) protein [3]. Here we present an unusual case of Triple A syndrome in whom neurological manifestations preceded adrenal insufficiency.
CASE REPORT
An 18-year-old boy was referred to the endocrine centre at the Sher-i-Kashmir Institute of Medical Sciences, Kashmir for the evaluation of suspected adrenal insufficiency. He was born full term, from a consanguineous marriage should be omitted to a native Kashmiri couple. His younger brother had died at the age of 1 year with possible adrenal insufficiency and his two other younger siblings were healthy (Fig. 1).
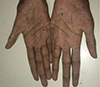
The patient reported generalised weakness, asthenia, fatigue, anorexia and progressive hyperpigmentation of skin for 6 months. Examination revealed a lean build patient (body mass index, 12.64 kg/m2), with hyperpigmentation of tongue, buccal mucosa and skin and a nasal twang to voice. His blood pressure was 100/60 mm Hg with postural drop. Neurological examination revealed wasting of thenar and hypothenar muscles (Fig. 2). The deep tendon reflexes were hyperactive and the sensory examination was unremarkable.
Baseline investigations revealed normal complete blood counts, serum creatinine and normal electrolytes (serum Na, 142 mmol/L; serum K, 4.1 mmol/L). Basal serum cortisol (8:00 AM) was very low (0.41 µg/dL) and failed to increase after intravenous injection of 250 µg of tetracosactin (30 minutes and 60 values of 0.47 and 0.45 µg/dL, respectively). The plasma adrenocorticotropic hormone (ACTH) levels were markedly elevated (1,262 pg/mL; normal range, 9 to 52 pg/mL). A diagnosis of primary adrenal insufficiency was established. A contrast enhanced computed tomography (CT) of abdomen failed to visualize adrenals indicating atrophic adrenals. The tuberculin test was negative.
The patient had significant past history. He initially presented to ophthalmologist at the age of 8 years with absence of tears. Schirmer test was suggestive of alacrimia. At that time he had no other complaints and was prescribed artificial tears. Four years later he presented with dysphagia especially to liquids. Barium meal examination revealed persistent narrowing in the distal region of esophagus near gastroesophageal junction. A diagnosis of achalasia cardia was made which was managed with esophagomyotomy. He had again presented at the age of 15 years with severe muscle wasting of thenar, hypothenar muscles and small muscles of feet. Nerve conduction tests revealed axonal peripheral neuropathy.
On the basis of primary adrenal insufficiency (with normal mineralocorticoid balance), alacrimia, achalasia cardia, and peripheral neuropathy a diagnosis of Allgrove syndrome was established. We have not performed the genetic study about the mutation of AAAS gene. A negative tuberculin test together with absence of adrenal calcification on abdominal CT made tuberculosis unlikely as the basis for adrenal insufficiency in our patient. He was started on oral hydrocortisone tablets 15 mg after breakfast and 10 mg at 6:00 PM. Patient reported a striking improvement on follow up at 4 weeks.
DISCUSSION
The description of Allgrove syndrome is limited to case reports and thus prevalence is unknown. The molecular basis for this rare autosomal recessive disorder syndrome is the mutated AAAS gene, located on chromosome 12q13, that codes for ALADIN protein. Most of the many reported mutations produce a truncated protein [4].
The cardinal features of Allgrove syndrome are ACTH resistant adrenal insufficiency, achalasia and alacrimia. The earliest feature is alacrimia as was in our patient. Adrenal insufficiency and achalasia are usually manifested during first decade of life. Achalasia of the cardia occurs in about 75% of cases and in older children/adults it usually manifests as dysphagia especially for liquids. Adrenal insufficiency is also an early manifestation and manifests as severe hypoglycemic or hypotensive attacks during childhood which may lead to sudden death. The death of the younger sibling of our patient at the age of 1 year was possibly due to adrenal insufficiency as he had presented with recurrent vomiting and shock. Adrenal insufficiency in Allgrove syndrome results from a progressive disorder leading to hypofunction of the adrenals at a variable time after birth [5]. The adrenal insufficiency in Allgrove syndrome is a unique form of primary adrenal insufficiency with preservation of mineralocorticoid production. In our patient the documentation of normal electrolytes indicated normal mineralocorticoid production although we have not measured plasma aldosterone levels. The only other cause of primary adrenal insufficiency with preservation of mineralocorticoid production is familial glucocorticoid deficiency. Although most of the patients with Allgrove syndrome have preserved mineralocorticoid production, it may be impaired in 15% of patients [6].
A neurological syndrome including central, peripheral, and autonomic nervous system impairment is often associated with Allgrove syndrome; neurological manifestations appear at a later age when compared with other manifestations. Distal sensorimotor polyneuropathy is a common manifestation [7]. Our patient presented at the age of 15 years with distal sensorimotor polyneuropathy, 3 years prior to manifesting adrenal insufficiency. Autonomic neuropathy is also frequently associated with Allgrove syndrome but our patient did not manifest any clinical features of autonomic dysfunction. Other neurological manifestations include mental retardation, optic atrophy, amyotrophy, ataxia, dysarthria, hypernasal speech, dementia, parkinsonism, dystonia, and chorea.
Other features that have been reported in association with Allgrove syndrome include microcephaly, short stature, dysmorphic features (long narrow face, long philtrum, down-turned mouth, thin upper lip, and lack of eyelashes), palmar and plantar hyperkeratosis, osteoporosis, and long QT syndrome [8].
We emphasize that Allgrove syndrome is a multisystem disease and the cardinal manifestations may appear at any time from infancy to adulthood.

 XML Download
XML Download