

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Small heterodimer partner (SHP) is an orphan nuclear receptor that, unlike other nuclear receptors, contains a putative ligand binding domain but lacks a classical DNA binding domain [1]. SHP mRNA is predominantly expressed in the liver, but it is also expressed in the adrenal gland, spleen, small intestine [2], and pancreas [3]. SHP has also been shown to interact with other nuclear receptors and transcription factors [4].
The expression of SHP is regulated by several other nuclear receptors. The orphan nuclear receptor, steroidogenic factor 1 (SF-1) and its liver homologue, liver receptor homolog-1 (LRH-1) transactivate the SHP promoter [5]. To date, at least 5 SF-1 binding sites have been identified in the promoter region of SHP [5], and LRH-1 is essential for basal SHP expression. When SHP protein expression is elevated by LRH-1, SHP forms a heterodimeric SHP/LRH-1 complex and this complex inactivates LRH-1 thus reducing SHP expression, which is an established auto-regulatory negative feedback loop for SHP [6]. Farnesoid X receptor (FXR) is another well-known inducer of SHP gene expression; an FXR binding site is located in the SHP promoter region [7]. In addition, liver X receptor α (LXRα) and hepatocyte nuclear factor 4α (HNF4 α) directly regulate SHP promoter activity, and their respective binding sites have been previously identified [89]. Other nuclear receptors and inducers, such as estrogen receptor-related receptor γ and sterol regulatory element binding protein-1 (SREBP-1), have also been known to regulate SHP expression [4].
In the liver, SHP is known to perform several metabolic functions. It regulates bile acid synthesis and cholesterol metabolism by modulating cholesterol 7α-hydroxylase (CYP7A1) expression through inhibition of LRH-1 and HNF4α activity [10]. In addition, SHP regulates lipogenesis by inhibiting SREBP-1c expression via LXR [11], and serum triglyceride levels by repressing microsomal triglyceride transfer protein (MTP) expression by inhibiting LRH-1 binding to the MTP promoter [12].
Thyroid hormone is known to regulate cholesterol and bile acid metabolism, mainly through regulation of 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase [13], low density lipoprotein (LDL) receptor [14], and CYP7A1 gene expression [15]. SHP has also been shown to play an important role in cholesterol metabolism [6] in the liver, mainly through transcriptional regulation of the CYP7A1 gene, which suggests a possible relationship or interaction between SHP and thyroid hormone. In our preliminary data using microarray analysis, we found that SHP expression was decreased by thyroid hormone.
Thyroid hormone acts mainly through its nuclear receptors, thyroid hormone receptor (TR) α and β. TRβ1 is putatively expressed in the liver [16]. The functional TR complex consists of a heterodimer with retinoid X receptor (RXR). This complex then binds to a thyroid hormone response element (TRE) and regulates gene expression [17]. Although we observed that thyroid hormone inhibited SHP expression, we were unable to identify TR binding to a TRE in the SHP promoter. However, TR could function through interactions with other nuclear receptors, like peroxisome proliferator-activated receptor (PPAR) and LXR [1618] at the level of transcription as well as by direct binding to TREs in the promoter region. Therefore, we speculated that TRβ might regulate SHP expression by interacting with other transcription factors that regulate SHP promoter. To confirm this hypothesis, we performed this study to find whether thyroid hormone regulates SHP expression by modulating the transcriptional activities of LRH-1, which is required for basal SHP promoter activity in the liver.
METHODS
Animals
C57BL/6J wild type mice were used. Mice were housed in groups of 4 or 5 in plastic microisolator cages at 22℃ with a 12-hour light/dark cycle. All animals were provided laboratory chow diet (Purina irradiated laboratory chow 38057, Purina Korea, Seoul, Korea) and water ad libitum. Animals were divided into 3 groups according to the duration of thyroid hormone treatment (n=4 or n=5 in each group). All experiments were performed in triplicate to confirm the results. Thyroid hormone, 3,5,3'-triiodothyronine (T3; Sigma Chemical Co., St. Louis, MO, USA) was prepared at a concentration of 1 mg/kg body weight in 20% DMSO and was administered via intra-peritoneal injection. Six hours group: T3 treated group was administered T3 6 hours before sacrifice. Five days group: the T3 treated group was administered T3 once daily for 5 days. Animals were sacrificed 24 hours after the last T3 injection. All animals were sacrificed after fasting for 6 hours beginning at 6:00 AM. Mice were anesthetized by an intraperitoneal injection of Zoletil (Virvac, Carros, France). Blood was drawn by inferior vena cava puncture. The liver was quickly removed, frozen in liquid nitrogen, and then used for RNA extraction. All procedures were performed in accordance with the guidelines of the Seoul National University Bundang Hospital Animal Policy and Welfare Committee.
Cholesterol measurement
Serum was prepared from whole blood by centrifugation at 1,200 ×g for 15 minutes. Serum total cholesterol was measured with an enzymatic assay kit (Thermo DMA Inc., St. Louisville, CO, USA) following the manufacturer's protocol.
Cell culture and transient transfection
Hepatocytes were isolated from wild type mice. Animals were anesthetized with Zoletil. The inferior vena cava was cannulated, and the portal vein was severed. Collagenase dissolved in Hanks' balanced salt solution (GIBCO Laboratories, Grand Island, NY, USA) was perfused through the liver at 37℃ to disperse hepatocytes, and then the hepatocytes were further purified by Percoll (Sigma). The cells were seeded into collagen-coated 6-well plates at density of 1 to 2×106 cells/well in 2 mL of William's E medium supplemented with 10% fetal calf serum, penicillin (100 U/mL), streptomycin (0.1 mg/mL), insulin (10 mg/mL), and triamcinolone (1 mmol/L). We also used HepG2 and HEK293 cells. Cells were grown at 37℃ in a humidified atmosphere of 5% CO2/95% air. HepG2 cells were maintained in modified Eagle's medium plus 10% fetal bovine serum (FBS). T3 (100 nmol/L) was incubated with primary hepatocytes and HepG2 cells according to the response time (control 2, and 6 hours). After T3 treatment, RNA was isolated from the cells.
One day before transfection, confluent cells were trypsinized and plated into 6-well plates at a 1:4 ratio to allow the cells to reach 50% to 60% confluency at the time of transfection. Six hours before transfection, fresh medium containing 10% charcoal-stripped FBS was added to the cells. All transfections were performed using the calcium phosphate method. Luciferase values shown are the averages of triplicate samples. All plasmids used in this study were previously reported. Various 5' deletions of the human SHP promoter [5], the human SHP promoter [19], LRH-1 [19], TRβ [20], and RXRα [21] were used.
RNA isolation and quantitative real-time polymerase chain reaction
Total RNA was isolated from frozen liver samples and cells using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according the manufacturer's instruction. First-strand cDNA was synthesized from 1 µg of RNA using superscript II reverse transcriptase (Invitrogen). Quantitative real-time polymerase chain reaction (PCR) was performed using SYBR Green PCR master mix and an ABI prism 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The PCR primers used for each gene are listed below (5' to 3'): mouse SHP-F (forward), TTG CAC CTG CAT CTC ACA GC; SHP-R (reverse), TCT TGG CTA GGA CAT CCA AG; glyceraldehyde 3-phosphate dehydrogenase (GAPDH)-F, TGT GTC CGT CGT GGATCT GA; GAPDH-R, CCT GCT TCA CCA CCT TCT TGA; human SHP-F, GGA CTT CCT TGG TTT GGA CA; SHP-R, CTC ATC CCA AGA AGG GAC AG; GAPDH-F, GAT CAT CAG CAA TGC CTC CT; GAPDH-R, TGT GGT CAT GAG TCC TTC CA. Relative mRNA expression was quantified using the comparative cycle threshold (Ct) method and expressed as 2-ΔΔCt.
Identification of differentially expressed genes
RNA was hybridized to Affymetrix GeneChip Human Gene 1.0 ST Arrays (Affymetrix, Santa Clara, CA, USA). Robust multichip analysis was used to normalize the expression data in the Expression Console software (Affymetrix) [22]. We calculated the log fold change of each probe in different conditions and independent samples. Student t test was performed to determine the significance of differential expression using GenePattern software [23].
Electrophoretic mobility-shift assay
Double-stranded oligonucleotides of the following sequences were used as probes: LRH-1 response element (LRE) in the human SHP promoter (LRE WT (-518 to -489), 5'-GAAGCA-GGGCCCAAGGTTAGGCAACAAG-3', LRE is underlined); LRE mutant competitor (LRE MT, 5'-GAAGCAGGGCCCttc-caaAGCAACAAG-3'); consensus LRE (5'-GACTTCTGGAGTCAAGGTTGTTGGGCCATTC-3'). The probes were labeled with [α-32P] dATP using Klenow polymerase (Roche, Mannheim, Germany), and the labeled probe (35,000 cpm) was incubated with nuclear extracts of HepG2 cells treated with 100 nmol/L T3 in 10 mmol/L Hepes (pH 7.9) containing 50 mmol/L KCl, 0.1 mmol/L ethylenediaminetetraacetic acid, 0.25 mmol/L DTT, 0.1 mg/mL poly (dIdC), 0.01% nonidet P-40, and 10% glycerol at room temperature for 10 minutes. Competitors were added in 50-fold molar excess to the labeled probe.
RESULTS
Thyroid hormone regulates the expression of genes involved in bile acid synthesis and cholesterol metabolism
Before conducting the microarray analyses, to confirm the effect of thyroid hormone on serum cholesterol, we measured serum cholesterol levels after intraperitoneal administration of T3 (1 mg/kg) to mice for 5 days. Serum total cholesterol levels decreased significantly after T3 injection (101.7±12.0 mg/dL vs. 47.0±9.7 mg/dL, P<0.05). We profiled the transcriptional changes in mouse liver before and after 5 days of T3 treatment and examined the expression of genes related to the regulation of cholesterol and bile acid metabolism.
In microarray analyses (Table 1), genes related to cholesterol metabolism, HMG-CoA synthase and reductase was up-regulated by thyroid hormone. Among genes associated with high density lipoprotein cholesterol, apolipoprotein A-1 was up-regulated and scavenger receptor was down-regulated by thyroid hormone. However, the LDL receptor, the main receptor responsible for clearing cholesterol-laden LDL particles from the blood, was not significantly up-regulated by thyroid hormone, which is different from the results of previous studies [1424].
Among the genes related to bile acid synthesis, CYP7A1 and mitochondrial sterol 27 hydroxylase (Cyp27a1) were up-regulated after T3 treatment, although the fold change of Cyp27a1 was small. In a previous study, thyroxine enhanced CYP7A1 expression but not Cyp27a1 expression in rat hepatocytes when added to dexamethasone-containing medium [25].
The expression of CYP7A1 and SHP was regulated by several transcription factors, such as FXR, LRH-1, HNF-4α, and LXRα [426]. However, expression of these transcription factors did not significantly change in response to thyroid hormone in microarray analysis (Table 1). In contrast, the expression of SHP was markedly decreased by thyroid hormone; therefore, we assumed that thyroid hormone regulates SHP expression independent of other transcription factors, and might regulate A expression indirectly through changes in SHP expression.
To confirm this, we examined the changes in SHP mRNA level after T3 in mouse liver by real-time PCR. Level of serum T3 was significantly increased in 6-hour and 5-day group than control (231.4±42.4 and 310.2±65.8 ng/dL vs. 70.1±20.7 ng/dL). As expected, the expression of SHP decreased after treatment with T3 for 5 days (Fig. 1A).
To confirm the specific regulatory effects of T3 on the expression of SHP, an in vitro study was performed using freshly isolated primary hepatocytes and human hepatoma HepG2 cells. Hepatocytes from C57BL/6L mice were isolated, and T3 (100 nmol/L) or vehicle was added to the culture media. As shown Fig. 1B, the expression of SHP decreased at 2 and 6 hours after T3 treatment. In HepG2 cells, T3 also decreased SHP expression after 6 hours of T3 treatment (Fig. 1C). Although the time sequence differed, T3 decreased the expression of SHP in both human and mouse; therefore, we evaluated the mechanism underlying the regulation of SHP by T3.
TRβ/RXRα plus T3 decreases SHP promoter activity by interfering with LRH-1
We used full-length (~2 Kb) mouse and human SHP promoter constructs [5] to determine whether TRβ affects the activity of the SHP promoter. We cotransfected HepG2 cells with SHP promoters along with an empty or TRβ/RXRα expression vector and evaluated the effect of vehicle or T3 (100 nmol/L) administered over 24 hours on promoter activity. T3 treatment alone did not have a significant effect on mouse or human SHP promoter activity (Fig. 2A, B). When the SHP promoter was cotransfected with TRβ/RXRα in the absence of T3, SHP promoter expression increased. However, this increased activity decreased significantly after administration of T3 (Fig. 2A, B). This result suggests that unliganded TRβ/RXRα increases the activity of the SHP promoter and T3/TRβ/RXRα repressed activity of SHP.
Chromatin immunoprecipitation and high throughput sequencing (ChIP-Seq) was used to define genome wide TRβ binding sites in the liver [27]. In accord with the negative results of bioinformatics searches for conserved TREs in the human and mouse SHP promoters, no such sites were observed. Therefore, we presumed that the T3/TRβ/RXRα complex might interact with other SHP promoter transcription factors. When we transfected the SHP promoter along with FXR, HNF4, or LRH-1, SHP promoter activity was increased (data not shown). Among these transcription factors, LRH-1 induced SHP promoter activity most strongly. LRH-1 is an abundant transcription factor in the liver; therefore, we checked for the interaction between LRH-1 and the T3/TRβ/RXRα complex using the human SHP promoter construct (Fig. 2C). Interestingly, LRH-1 and unliganded TRβ/RXRα markedly increased SHP promoter activity; however, when we added T3, promoter activity decreased significantly (Fig. 2C).
We then tested the functional interaction of TR and LRH-1 using SHP promoter deletion constructs affecting LRH-1 binding sites [5]. To check the interaction, we used 3 fragments (-2080, -1490, and -175) of human SHP promoter in our study. As LRH-1 response element (LRE) in the human SHP promoter was located from -518 to -489, the activation of promoter by unliganded TRβ/RXRα/LRH-1 was decreased in -175 fragments and when we added T3, overall activity of human SHP promoter by TRβ/RXRα/LRH-1 was significantly reduced (Fig. 3).
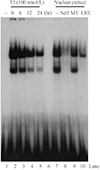
Electrophoretic mobility-shift assays were performed with a double-stranded oligonucleotide containing the -518 human SHP promoter LRE to explore the mechanism of the T3 dependent inhibition of LRH-1 transactivation (Fig. 4). The affinity of LRH-1 binding decreased in nuclear extracts of human hepatoma cells treated with T3 for increasing times up to 24 hours (lanes 2 to 5). To verify the specific binding of LRH-1 to the probe, competition assays were also performed. An oligomer containing a mutated LRE sequence did not compete with the probe (lane 9), whereas an oligomer with the consensus LRE competed with the SHP LRE probe (lane 10).
DISCUSSION
In this study, we demonstrated that liganded TRβ suppressed the expression of SHP. Because there is no known TRE in SHP promoter, liganded TRβ might decrease the expression of SHP by disturbing the transcriptional activity of LRH-1 to SHP. Therefore, our result could be one mechanism of regulation of CYP7A1 expression by thyroid hormone and TRβ.
In fact, role of thyroid hormone to regulation of CYP7A1 was already studied in several previous studies. Murine CYP7A1 is highly inducible by thyroid hormone because there are TREs in the murine CYP7A1 promoter [28]. Therefore, thyroid hormone directly increases the expression of murine CYP7A1 and induces bile acid synthesis. In human, there are inconsistent results about regulation of CYP7A1 expression by thyroid hormone. Previously, human CYP7A1 expression was reduced by thyroid hormone in several studies [2930]. Drover et al. [30] found that, unlike the CYP7A1 promoter site II TRE, the sequence of the site III TRE is not conserved among different species, which might explain the divergent responses of human and murine CYP7A1 to thyroid hormone. In contrast, recent study showed that thyroid hormone increased expression of CYP7A1 similarly in human and mouse primary liver culture, human hepatocytes and mouse liver [31]. However, it is difficult to explain until now why several researchers announced different results about thyroid hormone and induction of CYP7A1. For this reason, the relationship between CYP7A1 and thyroid hormone it is not sufficient to explain the regulation of bile acid metabolism by thyroid hormone. However, in our study, SHP expression in the mouse (both in vivo and in vitro) and human hepatoma cells was identically decreased by thyroid hormone. Therefore, our results are important to determine the mechanism underlying bile acid metabolism regulation, as it is the first study to report repression of SHP expression by thyroid hormone.
Many potential transcription factors for SHP, such as LRH-1, HNF-4, FXR, LXR, and SREPB-1 [4] were identified in previous studies. Among these, LRH-1 induces strong transcriptional activity because at least 5 LRH-1 binding sites were identified in the SHP promoter [5].
The results of ChIP-Seq, TRE bioinformatics and direct cotransfection all indicate that TRβ/RXRα does not regulate the SHP promoter directly. Among a number of transcription factors that are known to have such direct effects, LRH-1 has most binding sites and is essential for basal SHP promoter activity. In cotransfections of LRH-1 and TRβ/RXRα, we observed a T3-dependent suppression of promoter activity. This effect was diminished in -175 fragments of human SHP promoter. We also observed a time-dependent decrease in specific LRH-1 DNA binding in T3 treated livers, but the basis for this effect remains to be determined.
Thyroid hormone can act as both a positive and negative regulator of gene expression. In positive regulation, TR binds to TREs located in the promoters of target genes. In the absence of T3, the unliganded TR/RXR complex with corepressors inhibits the transcriptional activity of target genes. Binding of T3 to TR induces structural changes and recruits coregulatory proteins, which enables transcriptional activation [17]. TRs also negatively regulate transcription with and without DNA binding. In contrast to positive regulation, when TR binds to specific negative TREs, liganded TR recruits a corepressor complex to repress transcriptional activity. Another model suggests that TR does not bind to DNA directly, but instead binds to another transcription factor via protein-protein interactions. Then, liganded TR recruits a corepressor complex to inhibit transcriptional activity [32]. In contrast, unliganded TR binds to specific transcription factor and recruits a coactivator complex (CoA) leading to gene activation. The increased activity of SHP promoter by adding LRH-1 to unliganded TRβ/RXRα as shown Fig. 2C could be explained by this mechanism, although it is not clearly known yet from our results.
In our study, we assumed that TRβ did not bind to DNA directly, but interacted with another transcription factor, LRH-1. This interaction of TR with to other transcription factors has been demonstrated in previous studies; both TRβ and LXRα heterodimerize with RXRα and compete for binding to DR4 elements in the CYP7A1 promoter [16]. In addition, PPAR selectively inhibits the transcriptional activity of TRs by competing for RXR and possibly non-RXR TR-auxiliary proteins [18]. Therefore, transcriptional regulation by TR through interaction with other transcriptional factors might be as important as regulation through classic TREs.
In conclusion, we found that thyroid hormone indirectly regulates the expression of SHP mRNA by interfering with the transcriptional activity of another transcription factor, LRH-1. This provides an additional mechanism to better understanding of the regulation of bile acid and cholesterol metabolism regulation by thyroid hormone.




 XML Download
XML Download