

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Idiopathic congenital hypogonadotropic hypogonadism (CHH) is a rare reproductive disorder that is primarily caused by a gonadotrophin-releasing hormone (GnRH) deficiency but with significant genetic heterogeneity. Clinically, this disorder is characterized by abnormally low plasma levels of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) in conjunction with low or undetectable concentrations of circulating sex steroids. In approximately 50% of cases, CHH patients also suffer from a reduced or deficient sense of smell (hyposmia or anosmia, respectively), which is then termed as Kallmann syndrome (KS) [12].
KS was first recognized in 1856 by Maestre de San Juan [1] who observed patients with defective olfactory structures and a microphallus. A few years later, Kallmann et al. [2] identified the hereditary nature of this condition. In the 1950s, De Morsier and Gauthier [3] further described the partial or complete absence of the olfactory bulb (OB) and its axons in multiple hypogonadal males. Since then, a variety of non-reproductive dysfunctions and developmental anomalies have been observed in association with hypogonadism. This review aimed to provide a brief overview of the genetics, molecular pathogenesis, diagnosis, and treatment of CHH and KS, with a particular focus on recent progress in the field.
THE HYPOTHALAMIC-PITUITARY-GONADAL AXIS
Multiple developmental and neuroendocrine signaling pathways regulate the ontogeny and homeostasis of the GnRH neurons including the production, secretion, and action of GnRH [4]. The development of the olfactory system and GnRH neurons are intimately connected with each other and are often modulated by common cell surface receptors and chemoattractant or chemorepellent molecules. During early embryogenesis, GnRH neurons originate from the neural crest and ectodermal progenitors within the olfactory placode outside of the brain and then migrate in close association with the growing axons of olfactory receptor neurons and terminal nerves. After penetrating through the cribriform plate, the GnRH neurons arrive in the hypothalamus, where they detach from the olfactory axonal guides, become non-motile, and then disperse further into the brain basal lamina before undergoing terminal differentiation [5]. In mice, this migratory process begins at embryonic day E10.5 and is completed by E17.5 [6]. The timed and coordinated expressions of various cell adhesion molecules, axonal guidance cues, and extracellular matrix proteins, along with different neurotransmitters, transcription factors, and growth factors that regulate the migration of GnRH neurons, have been documented [5].
Within the hypothalamus, functional GnRH neurons extend their axonal processes to the medial eminence through which pulsatile GnRH is secreted into circulation via the hypophyseal portal system. GnRH binds to GnRH receptor 1 (GnRHR1) on gonadotroph cells in the anterior pituitary and stimulates the synthesis and secretion of LH and FSH. Subsequently, LH acts on the testes to stimulate testosterone production, and LH and FSH act on the ovaries to induce estrogen production which, in turn, leads to steroidogenesis and germ cell production [4]. GnRH is temporarily secreted at 3 to 6 months postnatally, which is more evident in boys than girls and is sometimes called "mini-puberty." GnRH secretion then remains dormant until the onset of puberty, when its reactivation initiates secondary sexual maturation [4]. Therefore, the normal development and properly coordinated actions of the hypothalamic-pituitary-gonadal (HPG) axis are essential for GnRH pulse generation and normal reproductive function (Fig. 1).
The examination of a 19-week-old human aborted fetus with X-linked KS revealed that the GnRH neurons were arrested in a tangle above the cribriform plate [7]. Because the initial differentiation and migration of the GnRH precursor cells appeared to be normal, it was speculated that the subsequent axonal elongation, path-finding, and/or terminal differentiation processes might have been disrupted and prevented the GnRH neurons from reaching the forebrain [7]. It was also proposed that the defective targeting, innervation, and synaptogenesis of the axons of olfactory sensory neurons to the OB anlage might have caused OB dysgenesis in KS patients. Following this study, CHH and KS were defined as the partial or complete failure of sexual development secondary to the failed embryonic establishment of hypothalamic GnRH neurons, which causes deficits in the secretion of sex hormones from the anterior pituitary and gonads [4567].
GENETICS AND GENETIC TESTING
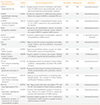
Recently, significant progress in genetic research regarding CHH has led to the identification of more than 31 different putative loci for this disorder, 17 of which are also associated with KS [8]. The current list of genes and information on their presumed biological activities and inheritance patterns are provided in Tables 1,2,3. Although these genes may be implicated in the etiology of approximately 50% of CHH/KS cases, the mutations in each of the genes account for less than 10% of such cases; furthermore, the majority of the underlying mechanisms have yet to be fully characterized [910].
CHH/KS may present as either a sporadic or a familial case following autosomal dominant, autosomal recessive, or X-linked recessive modes of inheritance. The variable phenotypes among individuals carrying the same mutation within a pedigree, even between monozygotic twins [11], have led to proposals of digenicity or oligogenicity, in which mutations of multiple genes synergize to produce a more severe phenotype but contribute to the phenotype with a variable penetrance [12]. For instance, loss-of-function mutations of semaphorin 7A (SEMA3A) have been reported in KS patients, but monoallelic mutations of this gene are not sufficient to cause the disease phenotype [13]. Thus, mutations of SEMA3A likely contribute to the pathogenesis through synergistic effects with mutations in other genes. Heparan sulphate 6-O-sulfotransferase 1 (HS6ST1) mutations account for approximately 2% of CHH cases and have been identified in combination with fibroblast growth factor receptor 1 (FGFR1) mutations in KS patients [14]. Additionally, mutations of prokineticin receptor-2 (PROKR2) in combination with mutations of anosmin-1 (ANOS1) [15] and FGFR1 [16] have been identified as well as potentially digenic patterns of nasal embryonic luteinizing hormone-releasing hormone factor (NELF)/ANOS1 and NELF/tachykinin receptor 3 (TACR3) mutations [17]. Patients with complex genetic makeups such as these are estimated to comprise at least 20% of all cases [10].
Establishing a phenotype-genotype association can aid phenotype-driven priority screening and better inform patient management and counseling [9]. Recent advancements in DNA sequencing technology have led to a wealth of genetic information regarding CHH/KS patients. In particular, the recognition of oligogenic traits [1012] has influenced perspectives on the genetic testing, diagnosis, and counseling for CHH/KS. For example, if an X-linked mode of inheritance is apparent, a mutation of KAL1 (note: following a nomenclature change by the Human Genome Organisation, this gene is now designated as ANOS1) is highly likely, especially when unilateral renal agenesis (30% to 40% of cases) and bimanual synkinesis (~75% of cases) are present [1819]. Kidney anomalies have not been associated with FGFR1 mutations, which is consistent with findings showing that the conditional knockout of FGFR1 in the mouse ureteric bud did not result in any kidney defects [20]. On the other hand, dental agenesis, cleft palate, and/or skeletal anomalies accompanied by varying degrees of hypogonadism with or without anosmia are indicative of a defective fibroblast growth factor (FGF) signaling pathway; thus, screening for FGFR1, FGF8, and HS6ST1 is recommended [1421].
If autosomal recessive inheritance is observed, alternations in GNRHR [22], kisspeptin receptor (KISS1R) [23], TACR3 [24], PROKR2 [25], or FEZ family zinc finger 1 (FEZF1) [26] can be suspected. If there are signs of CHARGE syndrome (Coloboma, Heart anomalies, Choanal atresia, Retardation of growth and/or development, Genital and/or urinary defects, and Ear anomalies and/or deafness), the chromodomain helicase DNA-binding protein 7 (CHD7) gene mutation should be considered a priority [27]. Mutations in dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1 (DAX1) cause CHH associated with adrenal insufficiency [28] and, if congenital deafness is present, then CHD7, SRY-related HMG-box gene 10 (SOX10), and/or interleukin 17 receptor D (IL17RD) can be suspected [2129]. CHH with obesity may indicate a mutation of leptin (LEP), leptin receptor (LEPR), or proprotein convertase-1 (PCSK1) [3031], whereas signs of other syndromic conditions, such as congenital ichthyosis [32] or spherocytosis [33], require that a comparative hybridization array or karyotype analysis be performed to detect any aberrant chromosomes.
THE EMERGING PICTURE OF MOLECULAR PATHOGENESIS
The developmental failure or misregulation of any one or combination of the genes and the signal pathways that are involved in GnRH migration, secretion, and/or activity at any stage will result in CHH and KS. For example, FGFR1 plays pleiotropic roles in human organogenesis including forebrain, skeleton, and neuroendocrine systems. The activity of this receptor is regulated not only by the binding of specific FGF ligands but also by alternative isoform expressions and other regulatory interactants. There is strong evidence to suggest that defects within the FGF signaling pathway underlie the pathogenesis of CHH/KS. Mutations in FGF8 (the ligand) and FGFR1 (the receptor) account for approximately 12% of CHH/KS cases [34]. Anosmin-1, a secreted extracellular matrix protein encoded by ANOS1, is the first mutated gene to be identified in KS patients [35]. Later it was shown that anosmin-1 binds to FGFR1 as a co-ligand and acts to fine-tune receptor signaling activity [36].
Heparan sulphate proteoglycans (HSPGs) are cell surface co-receptors that are essential for the formation of functional FGF/FGFR signaling complexes. The sequence specificity, length, and sulphation patterns of heparan sulphate are important regulators of receptor activity. Anosmin-1 may play a role in the recruitment of specific types of heparan sulphate to the FGF8/FGFR1 signaling complex [37]. In addition, mutations of HS6ST1, which is an enzyme that regulates the sugar modification of HSPG, have been identified in CHH patients [14]. More recently, mutations in other modulators of the FGFR1 signaling pathway, including FGF17, IL17RD, dual specificity phosphatase 6 (DUSP6), sprout homolog 4 (SPRY4), and fibronectin leucine rich transmembrane protein 3 (FLRT3), have been identified [21]; all of these were initially identified as part of the so-called FGF8 synexpression group.
The kisspeptin and tachykinin signaling systems also play key roles in the expression and release of GnRH at the time of puberty. Subpopulations of neurons that co-express kisspeptin and neurokinin-B are located in the arcuate and infundibular hypothalamic regions [38], and kisspeptin-producing neurons comprise the major afferents to GnRH neurons, which act to regulate the tonic feedback control of GnRH and/or gonadotropin secretion as well as the pre-ovulatory surge [39]. However, during development, signaling via kisspeptin and its receptor GPR54 are not necessarily required for GnRH neuronal migration per se [40]. Signaling mediated by tachykinin-3 (TAC3), which is the precursor of neurokinin-B, and TACR3, which is also known as neuromedin-K receptor (NKR), are also important for the metabolic regulation of GnRH neurons [38].
Because reproduction requires an adequate energy supply, the metabolic status is important for the regulation, stimulation, and homeostasis of GnRH neurons and gonadotrophs. In animal models, the insulin and leptin signaling pathways stimulate the reproductive endocrine system and regulate GnRH neuronal function [41]. It has also been shown that kisspeptin-expressing neurons are sensitive to changes in leptin concentrations and mutations in LEP or LEPR, result in CHH [30]. However, GnRH and kisspeptin neurons do not express the leptin receptor [41]; therefore, the mechanism by which leptin signaling can be detected and transmitted to kisspeptin/GnRH neurons remains unclear.
Although the molecular mechanisms of many of these putative genes remain under investigation, they can be divided into four categories according to their presumed biological activities: (1) development of craniofacial and forebrain structures; (2) embryonic migration of GnRH neurons; (3) expression and secretion of GnRH; and (4) maintenance and homeostasis of GnRH neurons and gonadotrophs. These divisions are not mutually exclusive because the associated events are often interdependent and multiple signal pathways cross-talk with one another. Ironically, genes that affect human reproduction should not be conserved in evolution. Nonetheless, a recent study revealed that founder mutations in four known KS/CHH genes (GNRH1, TAC3, PROKR2, and GNRHR) are shared by multiple unrelated individuals and that one founder allele of PROKR2 originated 9,000 years ago, implying there might be an advantage associated with this ancient mutation [42].
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
Although it is a relatively rare disorder, CHH/KS is the most common form of gonadotrophin deficiency, and it is about five times more prevalent in males. The reported frequencies vary across studies, ranging from 1 in 8,000 men and 40,000 women to 1 in 29,000 men and 130,000 women [4344]; ORPHA suggests that an average figure of 3.75 in 100,000 people is appropriate (www.orpha.net). A diagnosis of KS is often difficult to make due to genetic heterogeneities and the broad spectrum of phenotypic presentations. Patients with CHH are commonly diagnosed in late adolescence or early adulthood. Delayed puberty is classically defined as the absence of virilization and testicular enlargement (testicular volume <4 mL) in conjunction with the lack of or low sperm production by age 14 years in males and as primary amenorrhea and the absence of breast development (Tanner stage I) by age 13 years in females [45]. Any signs of eunuchoidal properties along with either anosmia or hyposmia should confirm the diagnosis of KS. When either a micropenis (5% to 10% of cases) or cryptorchidism (30% of cases) is present in infant males in conjunction with a lack of neonatal activation of the HPG axis at 3 to 6 months (mini-puberty), an early diagnosis of CHH can be made and then confirmed by the hormonal profiling of serum gonadotropins, sex steroids, anti-Müllerian hormone (AMH), insulin-like 3 (INSL3), and inhibin B [4647]. However, the occurrence of severe genital anomalies, such as hypospadias, likely suggests the presence of a human chorionic gonadotropin deficiency. Patients may also exhibit a variety of non-reproductive anomalies including craniofacial defects, such as cleft lip and/or palate and hypodontia, and eye defects, such as iris coloboma, ptosis, hypertelorism, and oculomotor palsy. Additionally, neurological symptoms, such as bimanual synkinesis, corpus callosum agenesis, and hearing impairments, and/or medical conditions, such as unilateral renal aplasia, may also be present [48].
Hypoplasia or aplasia of the OB and its tracts can be confirmed using high-resolution magnetic resonance imaging (MRI) but this does not always correlate with the clinical status of olfactory function; thus, a diagnosis of anosmia should be confirmed by formal smell testing and detailed questioning [49]. MRI scans are also useful during the CHH/KS examination to investigate the possible presence of any tumors or lesions in the hypothalamic and pituitary regions and to detect any problems in inner ear structures. To assess bone age and epiphyseal closure, bone densitometry can be used, whereas abdominal and/or pelvic ultrasonography aid in the detection of dysmorphisms in the kidney, ovaries, uterus, or testes [4348]. Low serum concentrations of FSH and LH, along with low testosterone (<3.7 nmol/L) and estradiol (<0.18 pg/mL) levels, are typically a consequence of decreased gonadotropin production [47]. Thus, a detailed assessment of anterior pituitary function must be undertaken to rule out other endocrinological conditions involving growth hormone and thyroid or adrenal functioning. Hyperprolactinemia may result in acquired HH (AHH) and, therefore, normal prolactin levels should also be confirmed [50].
Other complex syndromes present with clinical features that are similar to those of CHH or KS; these include combined pituitary hormone deficiency, septo-optic dysplasia, CHARGE syndrome, adrenal hypoplasia congenita with HH, Waardenburg syndrome, Bardet-Biedl syndrome, Gordon Holmes syndrome, Morning Glory syndrome, Hartsfield syndrome, and Dandy-Walker syndrome [8]. However, the most challenging differential diagnosis is that of constitutional delay of puberty and growth (CDPG). In the absence of anosmia, hormone profiling is not always fully informative regarding the confirmation of a diagnosis, although the combination of GnRH and human chorionic gonadotropin stimulation tests might help [51]. Signs that support a diagnosis of CHH include mild pubic and axillary hair growth due to the presence of adrenal androgens and the lack of a pubertal growth spurt while exhibiting steady linear growth. Thus, normal growth but longer arms and legs due to prolonged epiphyseal development may be suggestive of CHH, whereas individuals who are shorter than normal but have a proportional stature and delayed skeletal maturation while showing poor growth velocity are more likely to have CDPG [50]. Unlike CHH/KS, puberty is spontaneously initiated and eventually completed in patients with CDGP. Moreover, the presence of a micropenis or cryptorchidism, a cleft palate, sensorineural deafness, and anosmia are not observed in patients with CDGP. Nonetheless, no conclusive differential diagnostic tests are currently available for the differentiation of CHH/KS from CDPG.
Patients with CHARGE syndrome have both hypogonadism and OB aplasia/hypoplasia and can also share additional traits with KS patients, including a cleft lip or palate [52], which makes distinguishing this condition from KS extremely difficult. Potential systematic causes should also be eliminated prior to the confirmation of the diagnosis. Celiac disease, eating disorders, excessive exercise, and chronic diseases of the liver and/or kidney can all lead to AHH. The possibility of hemochromatosis, pituitary adenomas, and/or brain tumors should also be considered [8]. For further details regarding the diagnostic clues of CHH/KS, refer to a previously reported case series [48].
TREATMENT AND MANAGEMENT
CHH/KS can be managed more effectively if it is recognized during the early stages. The initial assessment of this disease typically relies on the physical examination and data obtained in primary clinics. Because these patients are often admitted by a variety of different clinics (endocrinology, pediatrics, neurosurgery, urology, obstetrics, and gynecology), a correct initial diagnosis and subsequent referrals are critical for appropriate follow-up visits and the systematic management of the disease on a long-term basis. Timely treatment to induce puberty can be also crucial for sexual, bone, and metabolic health and for the minimization of the psychological effects that can be associated with CHH/KS. The treatment regimen for CHH is principally determined by whether the goal is to develop secondary sexual characteristics (virilization or estrogenization) or to induce fertility as well. In most cases, fertility can be induced under specific regimens of testosterone treatment in males and estrogen/progesterone treatment in females, but only a few predictors have been identified [8]. In cases where gonadotrophs retain their functional secretory capability, the administration of exogenous pulsatile GnRH invariably induces the phasic release of LH and FSH and can restore puberty and fertility [50]. Patients typically require lifelong treatment to maintain normal sexual function, yet some patients sustain function of the gonads and remain fertile after testosterone withdrawal, which is called as "reversible" case [53]. Reversal of CHH may be more prevalent than appreciated. According to a retrospective study, the estimated lifetime incidence of the spontaneous recovery of reproductive function might be as high as 22% [54]. Interestingly, in these reversible cases, there is an enrichment of neurokinin B mutations, and the absence of the OB does not hinder the reversal. On the other hand, a subsequent relapse can also occur and, therefore, these patients require ongoing monitoring for reversal and/or relapse [54]. However, no predictors are currently available.
Recently, inhibin B, INSL3, AMH, and kisspeptin have emerged as potential biomarkers for the diagnosis and treatment of CHH [454647]. Kisspeptin may also be a potentially useful therapeutic target since a male patient reversed his condition after receiving exogenous kisspeptin infusions [45]. Although earlier studies indicate that the neonatal administration of gonadotropins could be beneficial [4450], further investigation is necessary to confirm its value for fertility outcomes and long-term effects. A comparative assessment of gonadotropin treatment during adolescence vs. adulthood and the benefits of prior treatment with androgens or FSH before pubertal induction are still under consideration [50].
Many studies have focused on testicular growth and gametogenesis in males [4647], but the rate of pregnancy in females with CHH/KS has yet to be sufficiently investigated, although this may be partly due to rarity of the disease and the long-term follow-up assessments required for gathering such information. Patients with cleft lip and/or palate, hearing loss, skeletal defects, or cryptorchidism require surgical and specialist interventions early in life. CHH/KS patients also have an increased risk of metabolic problems such as type 2 diabetes [55]. In females, low estrogen levels can result in osteopenia/osteoporosis [50] and, thus, bone density should be monitored. Due to the congenital nature of the disease, patients with KS who exhibit anosmia or hyposmia do not complain of altered flavor perception [49]. However, due to the potential detrimental effects of KS on quality of life and mental health issues, patients with KS should be carefully advised of this disability.
It is also important to recognize the implications of fertility treatment in CHH/KS patients because it leads to the inevitable risk of passing on the mutated gene to the next generation; this is especially true for genes with an autosomal dominant inheritance that have a 50% chance of transmission. Thus, genetic counseling should be provided to enable informed medical and personal decisions that rest on a clear understanding of the risks involved in conception, which can be even more critical in the case of suspected oligogenicity [91050]. Potential complications that may arise during pregnancy in female patients with certain anomalies should also be carefully assessed.
The major challenges associated with the treatment and management of CHH/KS include the appropriate timing of the treatment and how to promote adherence to treatment, especially during the transitioning of young adults from pediatric to adult care because any gaps can have considerable consequences. According to the consensus of the European Consortium on GnRH biology (COST Action BM1105, http://www.gnrhnetwork.eu/), a structured transition is preferable. Nonetheless, medical treatment provides only some aspects of the overall solution as many of the patients are emotionally traumatized, particularly by psychosexual issues and low self-esteem [9]. Thus, targeted psychosocial interventions, counseling, and peer support should aid in the amelioration of some of these issues.
CONCLUSIONS
Advancements in next-generation DNA sequencing technology hold much hope for more accurate detection and diagnostic procedures, personalized genetic counseling and screening, and tailored treatments for CHH/KS patients. Furthermore, future international multicenter trials that include large patient populations should aid in the development of an organized consensus for optimal management plans and fertility treatment options for these patients. Predictions based on genotype-phenotype correlations that consider oligogenicity, the identification of novel biomarkers, and the development of targeted therapies might also become realistic possibilities. Additionally, recent findings on the reversibility and oligogenicity of CHH/KS with variable phenotypes have challenged the conventional beliefs and current dogma associated with congenital defects. CHH/KS was previously believed to be secondary hypogonadism due to the absence of GnRH function [4567], but many of the genes associated with CHH/KS are broadly expressed in the HPG axis. Thus, the impact of the mutations may not be limited to hypothalamic GnRH neurons but may also contribute to primary defects in the pituitary or gonads downstream of GnRH receptor. Although several genes that ensure proper gonadal development have been identified, their links with CHH/KS has not been established. Additionally, the reversibility of this condition underscores the plasticity of the GnRH neuroendocrine system and suggests that further research is necessary.
Over the years, the distinction between CHH and KS has become even more blurry, in part because the mutations of known CHH/KS genes have been identified in other congenital syndromes that include CHH features but are accompanied by different symptoms [8]. The assumption is that it may not be a simple "all-or-none" story but rather a complex scenario that involves multiple feedback mechanisms. A broad range of associated phenotypes found in multiple disorders with HH potentially indicate that these issues are caused by overlapping signal pathways with different tissue specificities. Therefore, the need to redefine these congenital disorders and revise the existing perceptions of them is becoming more apparent, and it will be necessary to implement stratified medical strategies for these patients in the future.



 XML Download
XML Download