

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disease caused by various genetic defects of the enzyme involved in cortisol biosynthesis in the adrenal glands and ranges from mild to severe according to the degree of enzyme dysfunction [1]. Twenty-one-hydroxylase deficiency, occurring in 1 of 16,000 births [1], is the most frequent type of CAH. However, 17α-hydroxylase/17,20-lyase deficiency of CAH is very rare [2]. Loss-of-function mutation of CYP17A1 gene, which normally activates 17α-hydroxylase/17,20-lyase, can result in defective P450c17 action and present as primary amenorrhea and abnormality of secondary sex characteristics in women and as pseudohermaphroditism in men. The deterioration producing cortisol in the adrenal cortex stimulates adrenocorticotropic hormone (ACTH) secretion in the pituitary gland, leading to hyperplasia of the adrenal cortex. Long-term overproduction of ACTH due to inadequately treated or untreated CAH rarely transforms to adrenal cortical adenoma or carcinoma [3456]. Herein, we present a case of a huge adrenal mass confirmed as adrenal cortical adenoma in CAH with 17α hydroxylase/17,20-lyase deficiency.
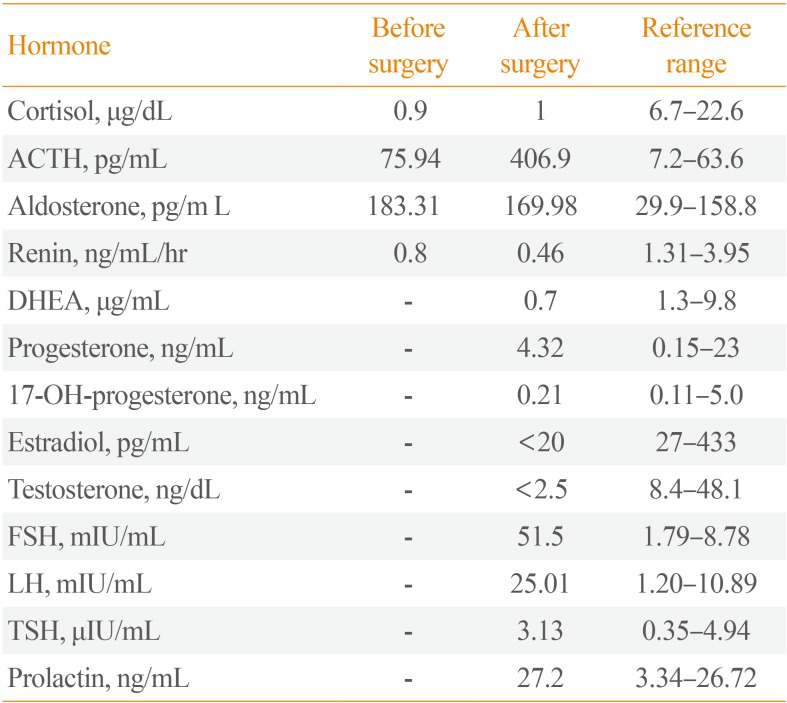
CASE REPORT
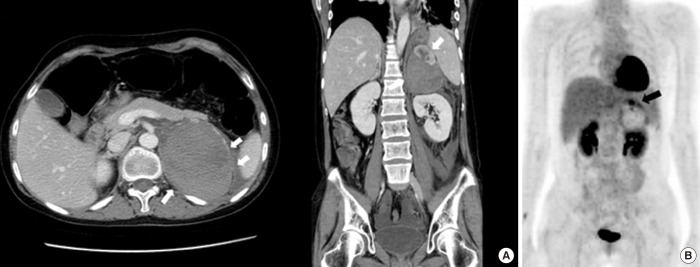
A 36-year-old female visited the emergency department with pain in the left upper abdominal quadrant. She brought her abdominal computed tomography (CT) scan taken in another hospital showing a 10×6.3×8.6 cm-sized left adrenal mass with hemorrhagic necrosis (Fig. 1A). On admission, her systolic blood pressure was 150 to 170 mm Hg and her diastolic blood pressure was 100 to 110 mm Hg, higher than the standard values for her age. She had no previous history of hypertension. Her weight was 63 kg, and her height was 175 cm. The routine biochemical study, random urine analysis, and blood coagulation test results were all normal, and the serum electrolytes showed Na 141 mmol/L (normal range [NR], 135 to 145), K 3.3 mmol/L (NR, 3.5 to 5.5), and Cl 100 mmol/L (NR, 98 to 120). Hormone tests showed cortisol was reduced to 0.9 µg/dL (NR, 6.7 to 22.6) and ACTH was elevated slightly, to 75.94 pg/mL (NR, 7.2 to 63.6). Renin was reduced to 0.80 ng/mL/hr (NR, 1.31 to 3.95) and aldosterone was elevated to 183.31 pg/mL (NR, 29.9 to 158.8). The aldosterone renin ratio was 22.9. Chest X-ray and electrocardiography were normal. The results of 24-hour urine analysis were as follows: metanephrine 0.518 mg/day (NR, 0 to 1.3), epinephrine 2.3 µg/day (NR, 0 to 20), and vanillylmandelic acid 4.04 mg/day (NR, 0 to 8); all values were within the NR except those for 17-ketosteroids 2.77 mg/day (NR, 7 to 20) and urinary free cortisol 37.6 (NR, 55.5 to 286). Typical CT findings of a large adrenal mass with hemorrhagic necrosis led to the diagnosis of pheochromocytoma with biochemically burnt-out status.
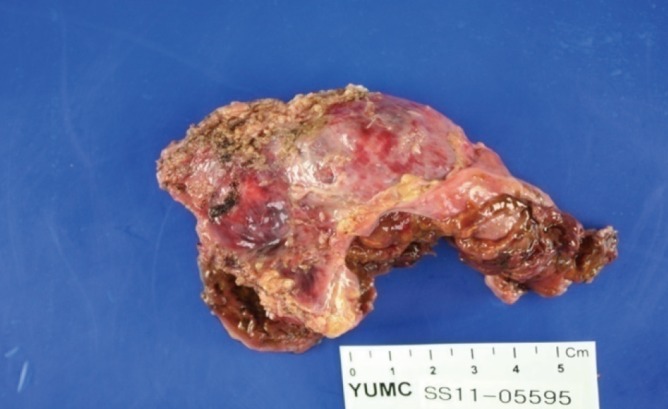
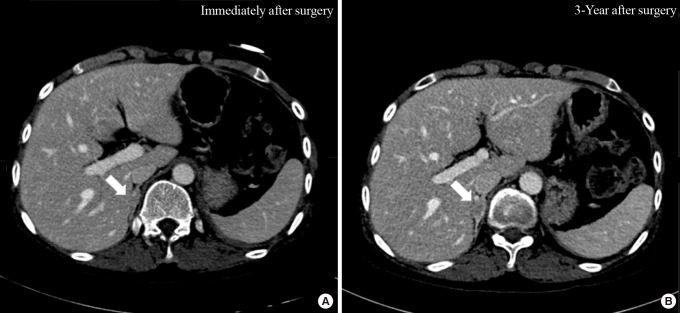
To further evaluate the characteristics of the adrenal mass, metaiodobenzylguanidine-single-photon emission computed tomography (MIBG-SPECT) imaging was performed and showed no significant radiotracer uptake. Next, 18F-fluorodeoxyglucose positron emission tomography-CT (18F-FDG PET-CT) was performed, and the FDG uptake in the solid portion of the mass was elevated (Fig. 1B). On CT scan, the contralateral right adrenal gland appeared relatively normal. The possibility of pheochromocytoma could not be excluded based on MIBG-negative and PET-positive findings. Thus, after proper premedication, the mass was surgically removed due its size. After laparoscopic adrenalectomy, a left hemorrhagic adrenal mass was pathologically confirmed as adrenal cortical adenoma 10×5×3 cm in size (Fig. 2). The patient complained of nausea and generalized weakness after the left adrenalectomy. Moreover, blood pressure remained at 190/119 mm Hg. After the operation, her ACTH was elevated to 406.9 pg/mL (NR, 7.2 to 63.6), and the low-dose ACTH stimulation test showed her cortisol was 1.0 µg/dL (NR, 6.7 to 22.6) at baseline, 1.6 µg/dL after 30 minutes, and 1.0 µg/dL after 1 hour showing blunted response. After reviewing her abdominal CT, we discovered that she had received plastic surgery on both breasts. We requested the patient regarding her medical history as she had failed to mention this procedure on the first visit. The patient had artificial prostheses inserted because she never developed breasts during puberty. She also lacked secondary sexual characteristics such as pubic and axillary hair (Tanner stage I). The patient confessed she experienced no menarche in puberty and had kept these facts secret even from her family members. Further hormone testing showed the followings: thyroid stimulating hormone 3.1 µIU/mL (NR, 0.4 to 4.9), prolactin 27.2 ng/mL (NR, 3.3 to 26.7), luteininzing hormone 25.0 mIU/mL (NR, 1.2 to 10.9), follicular stimulating hormone 51.5 mIU/mL (NR, 1.8 to 8.8), estradiol <20 pg/mL (NR, 27 to 433), progesterone 4.32 ng/mL (NR, 0.2 to 23), and 17-OH-progesterone 0.21 ng/mL (NR, 0.1 to 5.0). However, both dehydroepiandrosterone (DHEA) and renin were reduced to 0.41 µg/mL (NR, 1.3 to 9.8) and 0.7 ng/mL/hr (NR, 1.3 to 3.9), respectively, whereas aldosterone was elevated to 171.9 pg/mL (NR, 29.9 to 158.8) (Table 1). Furthermore, X-ray examination of the upper arm showed that the bone age was compatible with that of a 15-year-old female and bone mineral density was below the expected range for her actual age (Z-score -3.1 in the lumbar spine and Z-score -3.4 in the femoral neck), probably due to sex hormone deficiency. Chromosomal study of the patient was performed using peripheral blood leukocytes and showed a normal karyotype of 46, XX, female. We also conducted polymerase chain reaction for all intron sequences in the neighborhood of the CYP17A1 gene using appropriate primers. According to mutation analysis, we found a compound heterozygous mutation for p.Tyr329fs (c.985_987delTACinsAA) and missense mutation p.His373Leu (c.1118A>T) in exon 6. She was diagnosed with CAH caused by 17α-hydroxylase/17,20-lyase deficiency confirmed by the CYP17A1 gene mutation, presenting with lack of secondary sexual development, reduced production of cortisol and DHEA, increased aldosterone and ACTH as well as atypical huge adrenal mass. The patient was started on oral prednisolone, 7.5 mg in the morning and 5 mg in the afternoon, combined with estrogen. Finally, general weakness improved significantly, and the dose of oral prednisolone was reduced to 5 mg in the morning and 2.5 mg in the afternoon. To date, her hormone level has been monitored for 3 years. Due to her poor compliance and concerns of weight gain (although only 5 mg of prednisolone was prescribed), ACTH remained high. Inadequate suppression of ACTH resulted in the progression of hyperplasia of the right adrenal gland observed on the 3-year follow-up CT scan (Fig. 3).
DISCUSSION
CAH is characterized by adrenal enlargement resulting from excessive ACTH due to defective steroidogenesis. However, neoplastic transformation of the adrenal gland, such as adrenal cortical adenoma is a rare complication in CAH. Shimshi et al. [7] reported on a 51-year-old female with a 7-cm virilizing adrenocortical tumor in CAH due to 21-hydroxylase deficiency after reviewing nine cases including six adrenal adenomas and three carcinomas. Other studies reported adrenal masses diagnosed as adrenal cortical adenoma due to 21-hydroxylase deficiency [3457891011]. Kacem et al. [10] also reported on a 22-year-old patient with 11β-hydroxylase deficiency having a 16-cm-sized adrenal cystic mass. To date, most adrenal cortical adenomas or carcinomas reported in CAH have been due to 21-hydroxylase deficiency, and very few have been caused by 11β-hydroxylase or 3β-hydroxysteroid dehydrogenase deficiencies [12]. Although adrenal myelolipoma associated with CAH has been reported [13], reports on 17α-hydroxylase/17,20-lyase deficiency with a huge adrenal adenoma and degenerative cystic change in a patient have not yet been published.
The case in the present study involved CAH with 17α-hydroxylase/17,20-lyase deficiency and a compound heterozygotic gene mutation of p.Tyr329lysfs (c.985_987delTACinsAA) and p.His373Leu (c.1118A>T) at different sites on the same exon (exon 6), indicating that each mutation exists in different allelic genes as a heterozygote type. Cytochrome P450 17α-hydroxylase enzyme, an important enzyme involved in cortisol biosynthesis, is encoded by the CYP17A1, gene and 17α-hydroxylase/17,20-lyase deficiency is caused by a mutation in the CYP17A1 gene, which was first reported by Biglieri et al. [14] in a single patient [15]. CYP17A1 gene mutation resulting in 17α-hydroxylase/17,20-lyase deficiency deteriorates the synthesis of cortisol, testosterone, and estrogen, leading to the stimulation of ACTH and gonadotrophin hormone manifesting as hypergonadotropic hypogonadism, with accompanying overproduction of mineralocorticoids. Excessive mineralocorticoids may increase blood pressure or result in hypokalemia, as in this case. Moreover, due to the sex hormone deficiency, a patient with chromosome 46,XX will not experience secondary sex development and menstruation, whereas a patient with 46,XY will show female external genitalia [1617]. Primary amenorrhea, lack of secondary sexual development, and hypertension are the usual clinical presentations of 17α-hydroxylase/17,20-lyase deficiency. Atypically, the patient in this case came to the hospital at a comparatively old age complaining abdominal pain caused by the huge adrenal mass that had been suspected to be pheochromocytoma in the early stage, and this clinical manifestation was not the typical pattern compared with other CAH cases with 17α-hydroxylase/17,20-lyase deficiency. Kim et al. [18] reviewed the CYP17A1 mutations in adults with 17α-hydroxylase/17,20-lyase deficiency in Korea; however, clinical presentation of adrenal cortical adenoma in a patient has not been reported.
Adrenal enlargement and neoplastic change could be due to inadequate glucocorticoid treatment or non-treatment. Falke et al. [4] reviewed CT images in 13 cases of CAH according to the glucocorticosteroid treatment. Of the patients, seven who were treated properly showed no adrenal mass, but six, who were not treated properly showed gross enlargement of the adrenals. In our case, the patient had not been treated until her first visit at the age of 36; thus, the over 30-year continuous ACTH stimulation due to cortisol and sex hormone defects caused by 17α-hydroxylase/17,20-lyase deficiency led to the large adrenal adenoma. Whether the adrenal tumor was the result of long-term overstimulation of ACTH remains questionable. However, a correlation between adrenal hyperplasia and high ACTH levels is plausible given previous reports that adequate treatment that suppresses ACTH hypersecretion can reverse the adrenal enlargement [46].
Additionally, unlike the other cases of 17α-hydroxylase/17,20-lyase deficiency, the aldosterone level was high in our patient [14]. Yamakita et al. [19] suggested the severity of 17α-hydroxylase/17,20-lyase deficiency would be associated with the potency of cytochrome P450 corticosterone methyloxidase (aldosterone synthase), a key enzyme in the conversion of corticosterone to aldosterone. In severe 17α-hydroxylase/17,20-lyase deficiency, aldosterone synthase activity is more potentiated in outer zona fasciculata cells, leading to increased aldosterone. Furthermore, chronically elevated ACTH may have affected the production of aldosterone in zona glomerulosa, which is also responsive to ACTH [1920]. Of the three CAH cases with 17α-hydroxylase/17,20-lyase deficiency and a compound heterozygote gene mutation reported by Kim et al. [18], one patient with a high aldosterone level presented with higher blood pressure and ACTH level and lower cortisol (similar to our case) compared with the other two cases. Further studies of hyperaldosteronism-related phenotypes are necessary to explain the elevated aldosterone levels.
In conclusion, it is rare for CAH to manifest as adrenal cortical adenoma. Moreover, no huge adrenocortical adenoma has been reported in CAH with 17-α hydroxylase/17,20-lyase deficiency. In our case, a huge adrenal mass was initially considered a pheochromocytoma. Primary amenorrhea in female patients who present with both sexual infantilism and mineralocorticoid hypertension should be the focus of differential diagnosis. Additionally, proper glucocorticoid replacement is important for preventing adrenal gland enlargement or reducing the possibility of transformation into a tumor.
 XML Download
XML Download