

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Plurihormonality of pituitary adenomas is defined as the ability to produce more than one pituitary hormone, and the most frequently reported hormone combinations are growth hormone (GH), prolactin (PRL), thyroid stimulating hormone (TSH) and/or the α-subunit of glycoprotein hormones [1]. Acromegaly is one of the most common manifestations found in pituitary adenomas with plurihormonality [2]. However, the incidence rate and the clinical manifestations of the plurihormonal pituitary adenoma are still controversial. The anterior pituitary-specific transcription factor-1 (Pit-1), a transactivator of the GH, PRL, and TSH-β genes during anterior pituitary development, has been investigated as one of the possible candidates leading to the development of plurihormonality in these pituitary adenomas [34]. However, it has also been suggested that Pit-1 mRNA expression alone is not sufficient to determine plurihormonality in pituitary adenomas [5].
We describe a case of acromegaly caused by an invasive and plurihormonal pituitary macroadenoma. This patient also accompanied a rectal carcinoid tumor which was found not to contribute to the development of acromegaly. We also present the immunohistochemical findings of the pituitary adenoma and rectal carcinoid in order to confirm the plurihormonality of the pituitary adenoma and to investigate the presence of Pit-1 expression.
CASE REPORT
A 48-year-old woman was admitted to the department of endocrinology and metabolism in Kyung Hee University Hospital, complaining of gradually increasing size of both hands and feet and coarsely changing features of her face over the past 10 years. She had also experienced amenorrhea and a gradual weight gain of 13 kg for the past 10 years, with generalized myalgia but no headache, visual disturbance or galactorrhea. Her previous medical history included type 2 diabetes mellitus, hypertension and dyslipidemia. There was no specific familial or personal history. Height, weight, and body mass index were 163 cm, 62 kg, and 23.3 kg/m2, respectively. The vital signs were noted to be stable.
The sellar magnetic resonance imaging (MRI) revealed a large, 7.5-cm-sized sellar mass invading the supra-, infra- and parasella areas as well as the cavernous sinus. The mass extended to the level of the brain stem and compressed the optic chiasm. Though both the carotid arteries and cavernous sinus were encased by the mass, there was no sign of vascular compression or hydrocephalus. Moreover, the visual field examination also showed no defect. A colonoscopic examination which was performed to screen colon polyps, found a 0.4-cm-sized subepithelial nodule with an intact mucosa located approximately 5 cm above the anal verge and localized within the deep mucosa and submucosa.
Laboratory examinations showed hemoglobin A1c 7.1%. Insulin-like growth factor-I (IGF-I) level was 624 µg/L with a casual GH level of 16 µg/L. GH levels during a 75 g glucose loading test were all greater than 10 µg/L. TSH was 1.5 µU/mL, and PRL was 114.5 µg/L with follicular stimulating hormone (FSH) 1.8 mIU/mL, luteinizing hormone (LH) 0.2 mIU/mL and estradiol 47.7 pg/mL. TRH stimulation test demonstrated the paradoxical response of GH to TRH, and double stimulation test with insulin and GnRH to observe the remaining pituitary function revealed the blunted response of LH and FSH to GnRH (data not shown). These results confirmed the diagnosis of acromegaly with secondary amenorrhea due to hyperprolactinemia. In addition, it should be also noted that hypogonadism may be caused by the huge tumor size which resulted in compression of normal pituitary tissue.
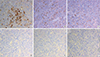
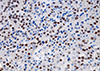
A transsphenoidal approach (TSA) was performed for the biopsy using the ring curettage and navigation system; acute complications did not occur. Light microscopy with hematoxylin and eosin (H&E) staining of the pituitary specimens revealed highly cellular tissues composed of monomorphic polyhedral cells with acidophilic cytoplasms and without mitoses. Immunohistochemical staining was performed using an automated immunostainer (Ventana Medical systems, Tucson, AZ, USA) according to the protocol of manufacturer with minor modifications as described elsewhere [6]. The antibody for Pit-1 was used. Tumor cells were demonstrated to be strongly positive for GH (Fig. 1A) and PRL (Fig. 1B), weakly positive for TSH (Fig. 1C), and negative for ACTH (Fig. 1D), FSH (Fig. 1E), and LH (Fig. 1F). Pit-1 was densely expressed in the nuclei of adenoma cells (Fig. 2). The expression of Ki-67, a cellular marker for cell proliferation, was found in less than 1% of the overall nuclear stained cells, and the tumor suppressor gene p53 were shown to be positive. Based on these findings, this pituitary adenoma case was immunohistochemically confirmed as possessing plurihormonality.
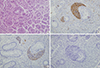
A subepithelial nodule on the rectum was completely removed with the endoscopic strip biopsy technique. Light microscopy with H&E staining of rectal specimen (Fig. 3A) revealed numerous monotonous cells having acidophilic cytoplasms and round to oval nuclei with nest-forming morphology around the normal intestinal glands. These cells were positive for CD56 (Fig. 3B) and synaptophysin (Fig. 3C), but negative for chromogranin (Fig. 3D). As a result, the subeipthelial mass obtained from the rectum was diagnosed as a carcinoid tumor, which was not accompanied by carcinoid syndrome with symptoms such as facial flushing, diarrhea or congestive heart failure.
Following TSA, the patient was started on monthly intramuscular injections of the long acting repeatable (LAR) form of octreotide 20 mg with daily bromocriptine 2.5 mg as an initial dose. However, GH levels remained higher than 10 µg/L, while IGF-I levels were measured between 350 and 400 µg/L even after increasing the dose to 30 mg of octreotide LAR. The annual follow-up MRI image demonstrated a gradual decrease in tumor size (Fig. 4).
DISCUSSION
According to the previous reports, the application of immunohistochemistry in diagnosing surgically removed pituitary tumors revealed that a certain number of pituitary adenomas appeared to be plurihormonal [17]. Though there is very little data on the exact incidence rate of such plurihormonal pituitary adenomas, the incidence rate is estimated to be about 10% to 15% of apparent pituitary adenomas [8]. Though the basic mechanism of plurihormonality has not yet been clearly revealed, one of the possible hypotheses is based on the role of various transcription factors such as Pit-1 [4]. Pit-1 gene is mapped to chromosome 3 and produces nuclear binding protein, which plays an important role as a transcriptional regulator [4]. Pit-1 belongs to the pituitary octamer unc-86 (POU)-domain family containing 60 amino acids in the POU-homeodomain region and 75-amino-acids in the POU-specific domain region. These two domains are joined together by the linker region and contribute to DNA binding [49]. Pit-1 is first expressed from embryonic day 13.5 and plays a major role in differentiation of the somatotroph, lactotroph, and thyrotroph cell lineages [410]. Cell-type specific and developmental state-specific transcription factors are implicated in the organogenesis of the anterior pituitary gland and subsequently produce five distinct cell types secreting six different hormones [11].
In this case, Pit-1 transcription was demonstrated in surgical specimens from the pituitary adenoma using IHC staining. Pit-1 in a mature pituitary gland has been reported to attribute to the over-expression of GH, PRL, GH/PRL, and TSH-producing pituitary adenomas, possibly leading to the functional differentiation and proliferation of tumor cells [812]. Altogether, it is possible to infer that plurihormonality of the pituitary adenoma is associated with Pit-1 expression. However, there has been no previous report investigating the relationship between Pit-1 expression and plurihormonality of pituitary adenomas. In addition, it has been reported that pituitary adenomas consistently expressed more Pit-1 mRNA than normal pituitary tissues [3]. Thus, it is feasible to speculate that Pit-1 expression in the tumoral state may have caused to dedifferentiate the cells to somehow more primitive stages and, as a result, a single cell may react to multiple hypothalamic hormones. A previous study by Yamada et al. [5] suggested that the presence of Pit-1 alone may not be sufficient to regulate hormonal production and tumor growth, and there could be other factors which play a role in determining the hormonal activity of pituitary adenomas. Thus, additional studies must be conducted to study the link between Pit-1 and the plurihormonality of pituitary adenomas.
It is well known that a large portion of GH-producing adenomas also produce PRL [13] as shown in our case. These adenomas usually behave more aggressively than pure GH-producing adenomas and are classified into three subtypes: the mixed GH cell/PRL cell adenomas, the mammosomatotroph cell adenomas and the acidophilic stem cell adenomas [1415161718]. The first two subtypes clinically present with acromegaly and mild hyperprolactinemia [13]. On the other hand, the acidophilic stem cell adenomas are characterized as demonstrating hyperprolactinemia but rarely acromegaly, and thus showing a distinct behavioral pattern of common prolactinomas [16]. Based on the main clinical feature of acromegaly, the huge adenoma in our case is likely to be either a mixed GH cell/PRL cell adenoma or a mommosomatotroph cell adenoma. The absence of the electron microscopy findings which is necessary to identify the exact immunohistologic subtype of mixed GH/PRL-producing adenomas is a limitation in this case.
It is well known that a large portion of GH-producing adenomas also produce PRL [13] as shown in our case. These adenomas usually behave more aggressively than pure GH-producing adenomas and are classified into three subtypes: the mixed GH cell/PRL cell adenomas, the mammosomatotroph cell adenomas and the acidophilic stem cell adenomas [1415161718]. The first two subtypes clinically present with acromegaly and mild hyperprolactinemia [13]. On the other hand, the acidophilic stem cell adenomas are characterized as demonstrating hyperprolactinemia but rarely acromegaly, and thus showing a distinct behavioral pattern of common prolactinomas [16]. Based on the main clinical feature of acromegaly, the huge adenoma in our case is likely to be either a mixed GH cell/PRL cell adenoma or a mommosomatotroph cell adenoma. The absence of the electron microscopy findings which is necessary to identify the exact immunohistologic subtype of mixed GH/PRL-producing adenomas is a limitation in this case.
One of the interesting points in this case is the lack of neurological symptoms in spite of its invasive nature. It has been reported that invasive adenomas were observed in 31% to 52% of plurihormonal adenomas, and some of plurihormonal adenomas are known to exhibit aggressive behaviors with poor prognosis [19]. One of the differences found in our case when comparing previous reports, Ki-67 index was <1%. It is very well known that invasive pituitary adenomas usually demonstrate higher Ki-67 proliferation index [20]. It has been not studied whether this low Ki-67 index may play a certain role in causing neurological symptoms in pituitary adenomas. The observation of no neurological symptoms in this case was unexpected considering its huge tumor size; and thus, this finding may merit further studies to clarify the factors which may influence on the occurrence of neurological symptoms.
Acromegaly caused by ectopic GH releasing hormone (GHRH) secretion or GH-producing tumors accounts for less than 1% of acromegalic patients [2122]. One of the key methods by which to differentiate ectopic acromegaly cases is by measuring the serum GHRH level [21]. Because the serum GHRH level was not measured, it was difficult to reject the possibility of GHRH production from the rectal carcinoid tumor and its consequent role in excessively stimulating GH production as well as the expansion of the pituitary macroadenoma. However, acromegalic features in this case developed very slowly over the course of 10 years, with the tumor size gradually increased to about 7.5 cm. When considering the very small size of the carcinoid tumor (0.4 cm) and its location, which was limited only to the mucosa and submucosal layer of the rectum, it is highly likely that the rectal carcinoid tumor was at a stage that was too early to produce enough GHRH to create a large long-standing pituitary adenoma. This is a very rare case of double primary tumors comprising a plurihormonal pituitary macroadenoma and rectal carcinoid tumor, which has not been reported in previous literature.
In this case, a 48-year-old female patient with acromegalic features was diagnosed as having a plurihormonal pituitary adenoma with concomitant rectal carcinoid tumor. Despite its large size and invasiveness, the patient did not have any neurologic symptoms. Based on the immunohistochemical staining of the specimens from the pituitary adenoma and rectal carcinoid tumor, it was assumed that these two tumors were neither clinically nor functionally related to each other. In order to elucidate the association between plurihormonality of the pituitary macroadenoma and the mutation of the Pit-1 gene, it would be necessary to extract DNA from both the pituitary adenoma and the rectal carcinoid tumor. Confirmation and comparison of the Pit-1 mutation and the DNA extractions would assist in determining whether or not these two tumors share an origin. In addition, further genetic studies are needed to determine whether other family members of this patient also demonstrate possible common pathways for formation of the pituitary adenoma and carcinoid tumor.

 XML Download
XML Download