

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Osteoclasts (OCs) are multinucleated myeloid lineage cells that form by cytoplasmic fusion of precursors that are formed in the bone marrow from myeloid lineage cells and circulate in the bloodstream [1]. These osteoclast precursors (OCPs) are attracted to sites on bone surfaces destined for degradation (resorption) in response to signals coming from these sites. These signals include receptor activator of nuclear factor-kappa B (NF-κB) ligand (RANKL), a multifunctional cytokine, which is expressed by a variety of cells in bone and bone marrow, including osteocytes, which are embedded in the calcified bone matrix, bone marrow stromal cells, and B and T lymphocytes [1]. Bone is continually being remodeled in the growing and adult skeleton in response to mechanical and other stimuli and to remove microscopic foci of damaged or effete bone. There are more than 1 million of these microscopic remodeling sites in the normal adult skeleton and the number increases in conditions in which OC formation is increased, such as sex-steroid deficiency, inflammatory bone disease, and hyperparathyroidism. In the first two of these conditions expression levels of pro-inflammatory cytokines, including tumor necrosis factor (TNF), interleukin-1 (IL-1) and IL-6 are increased and these, like parathyroid hormone (PTH), increase expression of RANKL to drive the increased osteoclastogenesis and activity [1]. RANKL interaction with its receptor, RANK, activates NF-κB signaling in OCPs and in OCs, which secrete protons, chloride ions, and collagenases from under the specialized ruffled portion of their cell membrane that faces the bone surface to be resorbed. Hydrochloric acid forms under this ruffled border membrane and dissolves the mineral component of the bone and cathepsin K is secreted to degrade the matrix [1]. OCs move along the bone surfaces in packs enlarging the resorption lacunae as they progress until the resorption process has been completed. Osteoblasts, the cells that form bone, are derived from mesenchymal precursors that, like OCPs, arise in the bone marrow and circulate in the blood from where they are attracted to resorption lacunae and attach to the resorbed surface and differentiate into osteoblasts. Most aspects of OC formation and activation are regulated by NF-κB signaling, which also limits OC formation induced by cytokines, including RANKL and TNF [1].
NF-κB SIGNALING PATHWAYS
NF-κB comprises a family of transcription factors, which positively regulate the expression of many genes involved in inflammatory and other responses by binding to their promoters. They were first identified as regulators of B lymphocyte differentiation that bound to the κB site of the κ light chain gene in B cells [2,3]. Subsequent studies showed that they were involved in innate and adaptive immune responses to pathogens and autoimmune stimuli and these established their critical roles in the initiation and maintenance of inflammatory conditions. However, they also regulate many aspects of normal cellular functions [2,3], and their activity is upregulated in many common conditions, including diabetes, atherosclerosis and cancer [4]. The NF-κB family includes RelA (also known as p65), p50, p52, RelB, and c-Rel. p50 and p52 are formed from larger precursor proteins, p105, and p100, which are encoded by NF-κB1 and NF-κB2, respectively [2]. All five family members have a Rel homology domain in their N-terminus that allows them to form homo- and heterodimers with one another and to bind to specific DNA sequences on gene promoters. DNA binding requires a C-terminal transcription activation domain, which RelA, RelB, and c-Rel possess, but p50 and p52 do not, and thus they rely on interactions with these 3 other family members to positively regulate gene transcription [4]. RelA and c-Rel preferentially form heterodimers with p50, and RelA/p50 activate most of the critical signaling in the canonical pathway, which occurs soon after activation is initiated. In many publications, NF-κB or NF-κB activity refers to canonical signaling mediated by RelA/p50. In OCs and many other cells, this occurs in response to cytokines, including RANKL, TNF, and IL-1 and is transient [5]. A non-canonical NF-κB pathway is activated several hours after canonical signaling has begun by translocation of RelB/52 heterodimers to the nucleus and is sustained, lasting for many hours. This activation occurs efficiently in response to RANKL, but not to TNF [6].
NF-κB signaling comprises a number of activation steps that require ubiquitination and proteasomal degradation or processing of proteins that function as inhibitors of signaling in cells under basal/unstimulated conditions by retaining NF-κB dimers in the cytoplasm of un-stimulated cells. These inhibitory NF-κB proteins are called IκBs. They include the canonical IκBs: IκBα, IκBβ, and IκBε [7], which have multiple ankyrin repeats that allow them to bind to NF-κB dimers and interfere with the function of their nuclear localization signals. RelA/p50 heterodimers are held in an inactivate state in the cytoplasm mainly by their interaction with IκBα, but they can also bind to IκBβ. RelA:RelA homodimers and c-Rel/RelA heterodimers preferentially bind to IκBε [4]. The C-terminal portions of p105, called IκBγ, and of p100, called IκBδ, also contain multiple ankyrin repeats, which endow them with IκB-like functions [8,9]. The IκBγ portion of p105 binds to RelA and c-Rel retaining them in the cytoplasm, but it can also bind to p50 molecules in RelA/p50 heterodimers [4]. Proteasomal processing of p105 occurs constitutively in un-stimulated cells and excises the C-terminal portion to generate p50 [10]. Upon stimulation by cytokines, such as RANKL and TNF, p105 is phosphorylated and rapidly degraded in the proteasome without release of p50. In addition, IκBα, which binds to RelA/p50 heterodimers, is degraded and this allows existing p50/RelA protein heterodimers to go to the nucleus. p100 also functions as an inhibitory protein in unstimulated cells when it is bound to RelB. Non-canonical NF-κB signaling leads to ubiquitination of p100, but instead of being degraded in the proteasome, it is processed to p52 and the resulting RelB:p52 heterodimers translocate to the nucleus. Interestingly, RelB also can function as an IκB and can bind to RelA to prevent it from activating canonical signaling [11].
ACTIVATION OF NF-κB CANONICAL SIGNALING
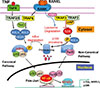
Canonical signaling is activated by a trimeric IκB kinase (IKK) complex, which consists of two catalytic subunits (IKKα and IKKβ) and a regulatory subunit, IKKγ, also called NF-κB essential modulator (NEMO) [4,12]. This IKK complex phosphorylates IκBα, leading to its polyubiquitination and degradation by the 26S proteasome and this is followed by translocation of RelA:p50 dimers to the nucleus (Fig. 1) [4]. Most of the IKK activity in the canonical pathway in cells, including RANKL-induced signaling in OC precursors, is mediated by IKKβ. Two important additional inhibitory effects of canonical IKK signaling are that it up-regulates: (1) early expression of IκBα, which initiates a negative feedback loop and limits subsequent RelA/p50 translocation [13]; and (2) p100 expression later to limit signaling in the non-canonical pathway [6]. As will be seen later, p100 has important inhibitory effects to limit RANKL- and TNF-induced osteoclastogenesis [14].
ACTIVATION OF NF-κB NON-CANONICAL SIGNALING
Non-canonical signaling is activated by IKKα following its phosphorylation by NF-κB-inducing kinase (NIK). In unstimulated cells, NIK is constitutively ubiquitinated on receptors, such as CD40, by TNF receptor-associated factor 3 (TRAF3) in a complex that includes TRAF2 and the inhibitor of apoptosis (IAP) proteins, cellular IAP 1 (cIAP1) and 2 [15]. Following stimulation by CD40 ligand in B cells, for example, cIAP1/2 ubiquitinate TRAF3, leading to its degradation and release of NIK from this complex. NIK then phosphorylates IKKα resulting in processing of p100 in the proteasome to p52 and the formation of RelB:p52 heterodimers (Fig. 1) [16,17]. The IκBδ portion of p100 binds preferentially to RelB to retain it in the cytoplasm, but interestingly it also binds to and regulates RelA homodimers [18].
Recent studies indicate that canonical [19] and non-canonical [20] NF-κB signaling negatively regulates mesenchymal precursor differentiation into osteoblasts, suggesting that NF-κB inhibitors should be able to stimulate bone formation. In addition, NF-κB is involved in certain aspects of endochondral ossification during skeletogenesis [21], but these aspects of NF-κB functions are beyond the scope of this review.
NF-κB SIGNALING IN OSTEOCLASTOGENESIS
A role for NF-κB in bone cells was first discovered unexpectedly in the mid-1990s when two groups of investigators generated NF-κB1/2 double knockout (dKO) mice in studies to further investigate the role of NF-κB in immunity and immune responses. They had already identified modest immune deficiencies in mice lacking NF-κB 1 or 2, RelB or c-Rel, while p65-/- mice died during embryogenesis from overwhelming TNF-induced liver cell apoptosis [22]. These dKO mice had failure of tooth eruption and osteopetrosis because they did not form OCs [23,24], but they also had severe B cell and T cell differentiation defects and did not form lymph nodes [23,24]. The osteopetrosis and immune deficiencies were reversed in the mice by transplantation of hematopoietic cells from wild type (WT) mice, indicating that the defects were of hematopoietic and not of mesenchymal cell origin [23]. The above OC, immune cell and lymph node formation defects were found also in RANKL and RANK knockout (KO) mice [5], which were generated a short time later independent of these studies. RANKL and RANK were discovered also largely unexpectedly by other investigators [25,26,27]. Although the NF-κB dKO and RANKL KO mice fail to form OCs, they have increased numbers of RANK-positive, TRAP-, cathepsin K-, and the calcitonin receptor-negative OCPs in their spleens [28]. These findings indicate that NF-κB signaling downstream of RANKL is not necessary for differentiation of myeloid cells into OCPs, but that it is required for terminal differentiation of these RANK-positive cells. Cytokines, including TNF, IL-1, IL-6, and RANKL did not rescue the defect in dKO OCP differentiation, indicating that NF-κB plays a central role in cytokine-induced OC formation [29]. Expression of RANK is promoted in myeloid precursors by macrophage-colony stimulating factor, which like RANKL is required for OC formation and regulates many aspects of OC formation and survival [30,31,32]. During RANKL-mediated osteoclastogenesis, RANK expression is transiently down-regulated by the β2 integrin, CD11b, to limit OC formation [33] during the earliest stages of OC formation.
NF-κB p100-/- and p105-/- mice have normal OC numbers and function in vivo, and IL-1 induces similar numbers of OCs from OCPs from these mice as it does from WT mice [29], indicating that neither is required for basal or IL-1-induced OC formation. RelA-/- mice die early during embryogenesis due to TNF-mediated massive hepatocyte apoptosis [22], but mice generated with deficiency of RelA only in hematopoietic cells have a defective response to RANKL in vivo [34]. Furthermore, inhibition of RelA nuclear translocation in OCPs in vitro inhibits osteoclastogenesis [34]. RelB-/- mice have near normal numbers of OCs, but their OCPs have an impaired response to RANKL in vitro [35]. No bone phenotypes have been reported in c-Rel-/- mice, and OCPs from these mice form normal numbers of OCs in response to RANKL in vitro [36].
CANONICAL NF-κB SIGNALING IN OSTEOCLAST FORMATION
Canonical NF-κB signaling is induced rapidly in OCPs in response to RANKL. For example, within an hour there is a rapid and transient increase in RelA and p50 mRNA expression levels [37]. During this period, RelA and p50 along with nuclear factor of activated T cells 2 (NFATc2; which, unlike NFATc1, is not required for OC formation) are recruited to the promoter of NFATc1, which has been called the master regulator of osteoclastogenesis, and this induces transient auto-amplification of NFATc1 expression [38]. The major role of NFATc1 at this very early stage of osteoclastogenesis may be to down-regulate expression of constitutively active repressors of RANK signaling [39], rather than induce expression of osteoclastogenic genes. These include Bcl6, which binds to the NFATc1 promoter in unstimulated OCPs and thus inhibits osteoclastogenesis. Bcl6 recruitment to the NFATc1 gene promoter is induced by CD11b during the early stages of osteoclastogenesis [33], and upon RANKL stimulation Bcl6 is replaced by NFATc1 to facilitate NFATc1 auto-amplification.
Interferon regulatory factor-8 (IRF8), Eos, and v-maf musculoaponeurotic fibrosarcoma oncogene family protein B are additional constitutively-expressed transcriptional repressors of RANK signaling in OCPs [39]. As will be seen later, negative regulation of signaling pathways in response to osteoclastogenic cytokines can play significant roles to limit OC formation and bone loss. The major role of RelA in OC formation is to prevent RANKL-induced OCP apoptosis, which is mediated by JNK, Bid and caspase 3 [34]. It remains to be determined how or if this is linked to the down-regulation of repressors of RANK signaling.
Following the transient increase in NFATc1 expression, c-Fos and p52 levels increase in OCPs ~2 hours after RANKL treatment and these remain increased thereafter through the later stages of osteoclastogenesis without any further change in RelA or p50 mRNA levels [40]. NFATc1 expression levels increase again 72 to 96 hours after RANKL treatment to induce expression of DC-STAMP, cathepsin K, TRAP, and other genes involved in OC resorptive functions. This increase in NFATc1 requires c-Fos expression induced by NF-κB since over-expression of c-Fos in NF-κB dKO OCPs induces NFATc1 expression and OC formation in the absence of RANKL stimulation [40]. Bcl6 also binds to the DC-STAMP and cathepsin K promoters in unstimulated OCPs and presumably is removed from these sites to facilitate NFATc1-induced OCP fusion and OC activation [41]. It has not been established if c-Fos expression is also required for the early transient induction of NFATc1 expression or why a more sustained expression of NFATc1 is not required to keep the above mentioned constitutively active repressors of RANK signaling inactive.
RANK, like other members of the TNF receptor superfamily, lacks intrinsic kinase activity to mediate downstream signaling [42]. In response to RANKL, RANK recruits a variety of molecules, including the multifunctional adaptor molecules, TRAFs 1, 2, 3, 5, and 6, and kinases, such as TGFβ-activated kinase-1 (TAK1), but of these only TRAF6 appears to be required for OCP differentiation in the canonical NF-κB pathway [42]. TAK1 induces activation of IKKβ leading to phosphorylation and subsequent activation of IKKβ, which phosphorylates IκB. The importance of this sequential activation process is highlighted by the observation that mice with deletion of IKKβ in OC lineage cells (IKKβf/f; CD11b-Cre [43,44] or IKKβf/f; Mx1-Cre mice [43,44]) have defective OC formation and osteopetrosis, and that OCPs from mice with a constitutively-active IKKβ (IKKβ-SS/EE) form OCs in the absence of RANK or RANKL treatment [45]. Expression of IKKβ, but not IKKα, is required for basal osteoclastogenesis and OC survival [43,44], and administration of a NEMO-binding domain peppeptide, which inhibits IKKβ activation, prevents OC formation and joint inflammation and erosion in mice with inflammatory arthritis [46]. Furthermore, macrophages, OCPs and immune cells deficient in IKKβ undergo apoptosis in response to TNF, an effect that is mediated by activation of JNK signaling [43,44]. Thus, increased IKKβ signaling mediates both joint inflammation and erosion in inflammatory arthritis.
NON-CANONICAL NF-κB SIGNALING IN OSTEOCLAST FORMATION
Several studies have reported that NIK-/-, p100-/-, and RelB-/- mice have normal numbers of OCs and no or minimal osteopetrosis in vivo [23,29,35,47], indicating that non-canonical signaling is not be required for basal OC formation. IKKα functions downstream of NIK in the non-canonical pathway, and it is not surprising therefore that IKKα-/- mice have normal OC numbers and bone volume. OCPs from IKKα-/- mice, similar to those from NIK-/- mice, do not form OCs in response to RANKL in vitro [47], but interestingly they form OCs in response to TNF or IL-1 [44]. In addition, mice lacking either NF-κB2 [23] or RelB [35] and NF-κB2/RelB dKO mice that we generated [48] have normal OC formation and bone volume in vivo. However, TNF induced similar numbers of OCs as RANKL from NF-κB2-/- OCPs [14], and RANKL induced fewer OCs from RelB-/- OCPs in vitro than from WT cells [35]. In addition, bone loss induced by cancer cells injected into tibiae of RelB-/- mice was significantly less than in WT mice [35], and overexpression of RelB can rescue the defect in OC formation from NIK-/- OCPs [49]. These findings suggest that non-canonical signaling mediated by NIK and RelB is required for the increased osteoclastogenesis induced by metastatic cancer in bone and in inflammatory arthritis. In contrast, we found that RelB-/- mice responded similarly to WT littermates and generated large numbers of OCs, associated with marked bone loss in response to daily injections of RANKL (unpublished observation). Metastatic cancers and inflamed joints produce a variety of factors in addition to RANKL, some of which can inhibit OC formation. In particular, RelB-/- mice have increased expression levels of several inflammatory cytokines and develop multiorgan inflammation as they age [50,51], indicting that the inflammatory milieu in the bone marrow of these mice is abnormal. Thus, the discrepancy between our findings with RANKL inducing brisk OC formation in the RelB-/- mice and these findings might reflect differences in the balance between stimulators and inhibitors of OC formation in the bone marrow microenvironment of RelB-/- mice with metastatic tumors or inflammatory arthritis.
NF-κB-MEDIATED NEGATIVE REGULATION OF OSTEOCLASTOGENESIS
Non-canonical NF-κB signaling is negatively regulated by TRAF3, and the CD40L-mediated degradation of TRAF3 by TRAF2/cIAP1/2 in the non-canonical pathway described above in B cells also occurs in OCPs in response to RANKL [14,52]. This results in NIK-mediated proteasomal degradation of p100 and p52/RelB nuclear translocation. TRAF3 is typically degraded in B cells through the ubiquitin-proteasome pathway by E3 ubiquitin ligases, such as Peli1 [53] and Triad3A [54], while deubiquitinases, such as OTUD7B, inhibit TRAF3 proteolysis and limit aberrant non-canonical NF-κB activation [55]. However, in OCPs, TRAF3 degradation occurs by autophagosomal, rather than proteasomal, degradation and its degradation is prevented in vitro by chloroquine [52], which raises the pH in lysosomes and thus inhibits lysosomal enzymes that function in acidic conditions [56].
Chloroquine has been used for many decades to treat malaria, and it and hydroxychloroquine are used also to dampen immune responses in inflammatory diseases, including rheumatoid arthritis and lupus erythematosis [57,58]. By preventing TRAF3 degradation, chloroquine dose-dependently inhibited RANKL-induced OC formation in vitro and prevented PTH-induced bone resorption and ovariectomy (OVX)-induced bone loss in mice in vivo [52]. TRAF3-/- mice die within the first week or two after birth with multiorgan inflammation associated with uncontrolled NIK activity, which is rescued by crossing the mice with p100-/- mice [59], consistent with p100 acting as a negative regulator of inflammatory signaling in the non-canonical pathway. This early lethality limits their use in study of the role of TRAF3 in OC formation and bone remodeling with age. To circumvent this hurdle, we generated mice with TRAF3 conditionally deleted in OC lineage cells and found that the mice developed osteoporosis as they aged as a result of increased OC formation and bone resorption [52], providing further evidence that TRAF3 functions as a major negative regulator of OC formation. These studies suggest that chloroquine could be administered to humans to prevent age- and sex steroid deficiency-related osteoporosis. However, chloroquine has a number of side-effects, which could limit its use in postmenopausal osteoporosis unless it could be targeted to bone.
RANKL and TNF activate NF-κB, c-Fos and NFATc1 sequentially in vitro in OCPs in a very similar manner [5,14,40], but TNF induces significantly fewer OCs in vitro than RANKL from WT OCPs [40]. TNF induces similar numbers of OCs from RANK-/- and WT OCPs in vitro, but it does not induce OC formation when administered to RANK-/- mice in vivo [27], suggesting that it induces one or more inhibitors of osteoclastogenesis. TNF and RANKL induce expression of the inhibitory NF-κB p100 protein [6], which is processed efficiently to p52 by RANKL, but not by TNF [60]. We found that TNF and RANKL induced similar numbers of OCs from p100-/- OCPs in vitro [14] and that TNF induced the formation of numerous OCs in p100/RANK dKO and p100/RANKL dKO mice in vivo, indicating that TNF can induce OC formation in the absence of RANKL signaling when this inhibitory protein is also absent [14]. Furthermore, TNF-transgenic mice lacking p100 developed more severe joint erosion and inflammation than TNF-Tg mice, providing evidence that p100 limits not only TNF-induced osteoclastogenesis, but also inflammation [14]. The importance of non-canonical signaling in TNF-induced inflammatory arthritis is further supported by the finding that NIK-/- mice have decreased joint inflammation in a model of TNF-induced arthritis [47]. These studies suggest that strategies to increase TRAF3 or p100 levels in OCPs and inflammatory cells should reduce joint inflammation and erosion in patients with TNF-mediated inflammatory arthritis.
Zhao et al. [39] have identified other TNF- and RANKL-induced inhibitors of OC formation. These include IRF8, which as mentioned earlier constitutively represses RANK signaling by inhibiting the expression and function of NFATc1 [61]. IRF8-/- mice have severe osteoporosis due to increased OC numbers and function, and OCPs from the mice form more OCs in response to TNF and RANKL than WT cells [61]. Recent studies report that IRF8 expression is down regulated epigenetically during RANKL-induced osteoclastogenesis through its DNA methylation by DNA methyltransferase 3a (Dnmt3a) [62]. These investigators found that RANKL induces a metabolic shift towards oxidative metabolism during osteoclastogenesis, accompanied by increased production of S-adenosylmethionine (SAM) a methyl donor in the methylation reaction, and that SAM-mediated DNA methylation of IRS8 by Dnmt3a represses expression of this anti-osteoclastogenic gene.
Another inhibitor of OC formation is recombination signal binding protein for immunoglobulin kappa J region (RBP-J) [63], which is the major transcriptional activator of Notch signaling. RBP-J suppresses induction of NFATc1 by dampening activation of c-Fos and suppressing induction of B lymphocyte-induced maturation protein-1 (BLIMP1) [63]. By these mechanisms, it prevents downregulation of IRF8 to maintain blockage of OCP differentiation. Mice deficient in RBP-J were protected from TNF-induced bone resorption and inflammatory bone destruction. In addition RBP-J expression is responsible in part at least for the osteopetrotic phenotypes in mice lacking either DAP12 or DAP12 and FcRγ [64] because the phenotype was largely rescued when these mice were crossed with RBP-J-/- mice and TNF was able to induce OC formation and bone resorption in DAP12-/- mice. RBP-J suppresses induction of NFATc1, BLIMP1, and c-Fos by inhibiting immunoreceptor tyrosine-based activation motif (ITAM)-mediated expression and function of PLCγ2 and activation of downstream calcium-CaMKK/PYK2 signaling. DAP12 and FcRγ mediate co-stimulatory signaling-induced OC formation downstream of ITAM. These important findings indicate that RBP-J suppresses ITAM-mediated co-stimulatory signaling and in this way limits crosstalk between ITAM and RANK/TNF receptors signaling. They highlight the complexity of inflammation-induced OC formation and activation and fine-tuning of osteoclastogenesis that is required during bone homeostasis and in inflammatory bone diseases.
 XML Download
XML Download