

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Langerhans cell histiocytosis (LCH), previously known as histiocytosis X, encompasses a spectrum of diseases with diverse clinical presentations and is characterized by proliferation and accumulation of pathological Langerhans cells in various organ systems [1]. The incidence of LCH is very low in the whole population and it is usually encountered in children aged 1 to 3 years at a rate of three to five cases per million people per year [2]. In particular, adult onset LCH is even rarer and its incidence has been reported to be around one to two cases per million people per year [3].
Generally, the choice of therapeutic regimen is based on disease severity. The International LCH Study of the Histiocyte Society proposes the stratification of LCH cases by the number of systems involved, and LCH is categorized into localized (single-system disease) and disseminated forms (multisystem disease) [4]. They further categorize those cases with single-system involvement by the number of sites within that system (e.g., monostotic vs. polyostotic bone disease; solitary vs. multiple lymph node involvement). In addition, the presence or the absence of risk-organ dysfunction is used to stratify patients with multisystemic disease; the presence of risk-organ dysfunction portends a poorer prognosis.
In adult LCH, presenting symptoms depend on the involved organs. Local pain (34%), particularly due to bone involvement, weight loss (11%), and fever (10%) are the most common symptoms at presentation and the most commonly involved organs are bone (57.3%) and lung (58.4%) [5]. Diabetes insipidus (DI) is the most common and permanent endocrine manifestation of LCH in adults and its prevalence is 29.6% [5].
Patients with localized disease are often successfully managed with local treatments like surgical resection, radiotherapy, and topical remedies [4]. For children with multisystem LCH, a number of single-center and multicenter randomized studies have shown the clear benefits of therapy with chemotherapeutic drugs and/or steroids [6]. However, a definitive treatment strategy for adult LCH has not yet been established and the Histiocyte Society launched the first international cooperative trial for the diagnosis and treatment of LCH in adults, known as LCH-A1 in 2004 [7].
We report the case of an adult patient who presented with central DI due to recurrent multisystem LCH involving the pituitary stalk and lung after surgery for localized primary LCH who was successfully treated with systemic steroids and chemotherapy, along with a literature review.
CASE REPORT
A 49-year-old man visited our hospital for polydipsia (8 to 9 L/day) and polyuria that started one month prior. He had no remarkable family history. Two years prior, the patient had experienced right chest pain and osteolytic lesions of the right 6th and 7th ribs were detected on X-ray. He was diagnosed with LCH after surgical excision of the rib mass in our hospital. At that time, subsequent bone scan and brain computed tomography (CT) revealed an osteolytic lesion in the occipital skull without involvement of brain parenchyma. The skull lesion was carefully followed up with skull X-ray, but the patient was lost to follow-up after 1 year.
On physical examination, the patient appeared well. Blood pressure was 130/90 mm Hg, pulse rate was 72 beats per minute, respiratory rate was 20 breathes per minute, and body temperature was 36.2℃. His height and weight were 164 cm and 70 kg, respectively. His tongue was not dry and skin turgor was normal without evidence of dehydration. Inspection and palpation of the chest revealed no masses, lung sounds were clear at auscultation, and no lymph nodes were palpable in the neck or limbs. Neurologic examination revealed normal motor and sensory functions, symmetric reflexes, and no evidence of clonus, fasciculations, or ataxia. The results of other physical examinations were unremarkable.
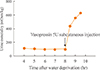
Blood cell count, urinalysis, serum chemistry, and electrolytes were within normal range. Basal anterior pituitary hormones were also normal. Serum osmolality at admission was 295 mOsm/kg and urine osmolality was 75 mOsm/kg. For differential diagnosis of polydipsia, the water deprivation test was performed. After 8 hours of water deprivation, serum osmolality reached 303 mOsm/kg and urine osmolality was stable with variation of less than 30 mOsm/kg, ranging from 99 to 118 mOsm/kg. We injected five units of vasopressin subcutaneously and urine osmolality was followed serially. After vasopressin injection, urine osmolality markedly increased to 562 mOsm/kg (more than 50% from baseline) after 1 hour and we reached a diagnosis of central DI (Fig. 1).
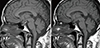
Magnetic resonance imaging (MRI) of the pituitary gland revealed a solid mass involving the infundibulum (6 mm) and the posterior lobe of the pituitary gland with loss of bright spot on T1 weighted imaging (Fig. 2A). We further performed the combined pituitary stimulation test and anterior pituitary function was normal.
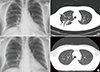
Chest X-ray showed cavitary lesions in the right upper lobe and reticular opacities in right lower lobe (Fig. 3A). CT of the chest with contrast enhancement demonstrated multiple patchy infiltrations in both lung fields with ground glass opacity and air bronchogram (Fig. 3B). Chest CT also revealed new osteolytic bone lesions in the right 9th rib and left 10th rib, which were visualized as hot uptake on bone scan.
We concluded that LCH had spread to the pituitary gland, lung and bone and we planned treatment with inhaled desmopressin and systemic chemotherapy. The patient received intranasal desmopressin 10 µg twice daily and urine volume decreased to 1.5 to 2 L per day. He was treated with 6-week induction chemotherapy with prednisolone (40 mg/m2/day for 6 weeks) and vinblastine (weekly 6 mg/m2 intravenous bolus injection), followed by maintenance chemotherapy with 6-mercaptopurine (50 mg/m2 per day), vinblastine (6 mg/m2 intravenous bolus injection every 3 weeks), and prednisolone (40 mg/m2/day for first 5 days every 3 weeks) for 12 months.
Two months later, follow-up sella MRI demonstrated disappearance of the mass in the infundibulum of the pituitary gland (Fig. 2B). During maintenance chemotherapy, chest CT with contrast enhancement revealed complete disappearance of the lung lesions 11 months after the start of chemotherapy (Fig. 3C, D). Six years after chemotherapy, the patient has shown no evidence of recurrence at regular follow-up examinations and polyuria is well controlled with intranasal desmopressin.
DISCUSSION
According to a comprehensive review of the 274 adult LCH patients aged 18 years or older from 13 countries enrolled in International Histiocyte Society Registry from January 2000 to June 2001, 68.6% presented with multisystem disease [5].
Clinical characteristics of LCH are as varied as organ involvement. In the case of pituitary gland involvement, DI is the most common endocrine abnormality, occurring in 12% of children with LCH and 30% of adults with LCH [8]. Anterior pituitary dysfunction is found in up to 20% of patients with LCH, and is almost always associated with DI [8].
Established DI is generally permanent [5,8]. One prospective study evaluating the efficacy of chemotherapy among 109 childhood LCH patients found that established central DI (eight of 109 patients) was neither reversed nor ameliorated by any treatment [9]. In another study evaluating the efficacy of chemotherapy in childhood LCH, preexisting central DI persisted after chemotherapy in 23 patients, with the exception of two cases of partial central DI in which the disappearance of pituitary stalk thickening was observed on MRI and desmopressin was no longer required (complete remission) [10]. This emphasizes the need for early intervention before DI is fully established. The pathogenesis of DI has been suggested to be infiltration or scarring of the hypothalamo-pituitary axis or an autoimmune process involving antibodies against antidiuretic hormone [11].
The high proportion of cases with lung involvement is the most conspicuous difference between adult and child LCH [5]. Symptoms of pulmonary involvement are cough, dyspnea, and chest pain, or the disease may be asymptomatic, as seen in our case [12]. The probability of survival at 5 years postdiagnosis is the lowest among isolated pulmonary disease patients (87.8%), compared to 100% in single-system disease and 91.7% in multisystem disease [5].
Chemotherapy options for LCH include combined treatment with vinblastine, etoposide, mercaptopurine, corticosteroids, azathioprine, cyclophosphamide, chlorodeoxyadenosine, and cytosine arabinoside [4]. Cooperative international efforts by the Histiocyte Society have produced a considerable amount of information about LCH in childhood and its treatment response [13,14]. The standard chemotherapy regimen for multisystem LCH in children has been established to be a combination of vinblastine and prednisolone administered over 12 months [14].
Unlike LCH in children, therapeutic regimens of adult LCH have not yet been confirmed, and complete or partial remission rates are not high (21% to 65%) [3]. The current chemotherapeutic regimen is determined at the physician's discretion based on case or case series reports of successful treatment. Therefore, the Histiocyte Society launched the first cooperative international trial for the diagnosis and treatment of LCH in adults in 2004. This trial, known as LCH-A1, seeks to define a common treatment strategy for patients with multisystem and pulmonary LCH and to evaluate the efficacy of the standard regimen (vinblastine and prednisone) for multisystem childhood LCH in adult LCH patients [7].
In Koreans, several cases of LCH involving the hypothalamo-pituitary axis or other organs have been reported [15,16]. Recently, Hong et al. [17] reviewed the clinical characteristics of 10 LCH cases with hypothalamo-pituitary involvement, and adult onset LCH patients were more likely to have anterior pituitary gland dysfunction and a poorer prognosis than childhood onset LCH patients. However, no studies have evaluated the efficacy of treatment regimens in Koreans.
Our case of multisystem adult-onset LCH presented with polyuria and was subsequently diagnosed with central DI due to pituitary stalk and lung involvement of LCH. Based on the Histiocyte Society's LCH-A1 study in adults, the patient was successfully treated with a combination of corticosteroids, vinblastine, and mercaptopurine without major adverse effects. The pituitary mass disappeared and lung lesions resolved after chemotherapy. Our successful treatment of multisystem adult LCH demonstrates that the LCH-A1 regimen is an effective and well-tolerated therapeutic regimen.
 XML Download
XML Download